Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
|
Cardiac & Cardiovascular Systems
|
Peripheral Vascular Disease
Inorganic phosphate is essential for a variety of cellular processes, such as energy metabolism, bone formation, and synthesis of biomolecules, including phospholipids and nucleic acids. However, elevated serum phosphorus has emerged as a key risk factor for vascular calcification.
- Phosphate
- Vascular Calcification
- Cardiovascular
- Calcium phosphate
1. Role of Phosphate
During the past decade, in vitro experiments have shown calcium-phosphate deposits in vascular smooth muscle cells incubated with high phosphate concentration [1]. Logically, this observation was first interpreted as the consequence of an increase in phosphate transport, with the consequent increase in the intracellular phosphate concentration [2]. However, several studies show that phosphate transporters are saturated with normal serum phosphate levels [3][4]. Moreover, additional studies show that the formation of calcium-phosphate crystals is a passive physicochemical process that does not require any cellular activity, suggesting an important role of phosphate homeostasis [5][4][6]. Notably, there are two major consequences regarding the fate of vascular smooth muscle cells in phosphate-induced vascular calcification. The first involves apoptosis-dependent matrix mineralization, which has been detected both in cultured human vascular smooth muscle cells and in arteries from pediatric dialysis patients [7][8][9]. The second invokes a profound transition to a bone-forming phenotype [10]. In support of this notion, in vitro studies have shown that elevated phosphate results in the expression of osteochondrogenic markers (such as BMP-2 and Runx2/Cbfa1, a transcription factor that induces the expression of major components of the bone matrix) [11][12]. However, recent studies show that calcium-phosphate deposits can induce both the transition to a bone-forming phenotype and apoptosis in vascular smooth muscle cells and the aortic wall, suggesting that the active mechanisms described could be a response to the excessive formation and deposit of calcium-phosphate crystals [13][5][14][15].
2. Biomineralization Process
The formation and deposition of inorganic minerals within or outside the cells of various organisms is known as biological mineralization or biomineralization. Biomineralization in hard tissues (such as in bone or dentine) is normally considered a physiological process; however, the accumulation of inorganic minerals in soft tissues (such as blood vessels, joints, and internal organs, including muscle, liver, or brain) is considered pathological or ectopic biomineralization. Under normal conditions, the soft tissues are not mineralized, but due to aging and other pathological conditions, soft tissues become calcified, which leads to morbidity and mortality. The main biominerals found in mineralized vertebrate connective tissue are calcium-phosphate salts. In an aqueous system of calcium and phosphate, there are several known non-ion-substituted calcium phosphates, which have also been found in calcified tissues. The phosphate ion is a central phosphorus atom surrounded by four oxygen atoms in a tetrahedral arrangement. In biological systems, it is found as a free phosphate ion in solut00000000000000000ion (inorganic phosphate) or bound with different biological molecules, including proteins, sugars, lipids, and nucleic acid. Aqueous inorganic phosphate exists in four forms according to its triprotic equilibrium: (1) trihydrogen phosphate ion (H3P04), (2) dihydrogen phosphate ion (H2PO4−), (3) hydrogen phosphate ion (HPO42−), and (4) phosphate ion (PO42−); (see Figure 1). Inorganic phosphate is quite strong with respect to the first dissociation (pKa1 = 2.1), moderately weak with respect to the second (pKa2 = 6.9), and very weak with respect to the third (pKa3 = 12.4). Under strongly basic or acidic conditions, the phosphate ion or trihydrogen phosphate dominates, respectively. In extracellular fluid (pH = 7.4), only H2PO4− and HPO42− ions are present in significant amounts in a proportion of 1:4, respectively. Whereas, in cytosol (pH = 7) and lysosome (pH = 4.8), this proportion is inverted (1.6:1 and 99:1, respectively).
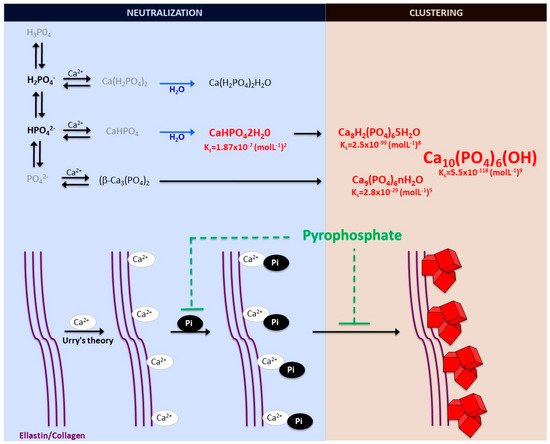
Figure 1. Schematic representation of calcium-phosphate crystal formation. Phosphate exists in four forms in biological systems: trihydrogen phosphate (H3P04), dihydrogen phosphate ion (H2PO4−), hydrogen phosphate ion (HPO42−), and phosphate ion (PO42−). Various phosphate-calcium salts are produced in the presence of calcium, including anhydrous monocalcium phosphate (Ca(H2PO4)2), anhydrous dicalcium phosphate (CaHPO4), β-tricalcium phosphate (β-Ca3(PO4)2), monocalcium phosphate monohydrate (Ca(H2PO4)2H2O), and dicalcium phosphate dihydrate (CaHPO42H20, also called Brushite). The final product of the calcium and phosphate reaction is crystalline hydroxyapatite (Ca10(PO4)6(OH)), the main component of bone and calcified tissues and two of its precursors, amorphous calcium phosphate (Ca9(PO4)6nH2O) and octocalcium phosphate (Ca8H2(PO4)65H2O). Pyrophosphate directly inhibits the formation and growth of phosphate-calcium crystals, mainly hydroxyapatite.
Notably, various phosphates-calcium salts are obtained by charge neutralizing these different inorganic phosphate ions in the presence of a calcium ion [16], including (1) monocalcium phosphate anhydrous (Ca(H2PO4)2), (2) dicalcium phosphate anhydrous (CaHPO4), and (3) β-tricalcium phosphate (β -Ca3(PO4)2) [17][18]. Both Ca(H2PO4)2 and CaHPO4 are hydrated to form their hydrated forms (monocalcium phosphate monohydrate (Ca(H2PO4)2H2O) and dicalcium phosphate dihydrate (CaHPO42H2O). CaHPO42H2O, also called Brushite, is often found in calcified tissues, whereas Ca(H2PO4)2, Ca(H2PO4)2H2O, CaHPO4, and β -Ca3(PO4)2 have never been found in calcifications [18]. The Mg-substituted β-tricalcium phosphate form (whitlockite) is not formed under physiological conditions. However, whitlockite was also found in some calcified tissues, such as in the aorta in hemodialysis patients [19][20].
The final product of the calcium-phosphate salts’ reaction in neutral or basic solutions is crystalline hydroxyapatite (Ca10(PO4)6(OH)), the main component of bone and calcified tissues; along with two of its precursors (amorphous calcium phosphate, Ca9(PO4)6nH2O, and octocalcium phosphate, Ca8H2(PO4)65H2O) [17][21]. Notably, amorphous calcium phosphate, which is also found in soft-tissue pathological calcifications, consists mainly of roughly spherical Ca9(PO4)6 clusters (called Posner’s clusters) that appear to be energetically favored compared to (Ca3(PO4) and Ca6(PO4)4 clusters [16][17]. Therefore, the structure of hydroxyapatite [21] can be interpreted as an aggregation of Posner’s clusters [22][23] (Figure 1). Notably, Mg2+ and ATP are critical for the stabilization of amorphous calcium phosphate [24][25].
According to the charge neutralization theory of calcification [26], the high glycine content of elastin and collagen proteins favors the formation of beta-turns that are known to interact with calcium ions. Therefore, the deposition of these calcium-phosphate salts, both in vitro and in vivo, takes place on these extracellular matrix proteins [13][5]. For example, in bone and connective tissues, these salts are predominantly deposited on the elastic and type I collagen fibers. Moreover, in the aorta wall, calcium-phosphate crystals are deposited on elastin, the main component of the elastic fibers in the medial layer [5]. Notably, a study showed that a mouse model of elastin haploinsufficiency exhibited a significant reduction in arterial calcification [27]. In contrast, phosphate-induced mineralization, both in vitro and in vivo, is accelerated by the products derived from elastin degradation [28].
Finally, the depositions of calcium-phosphate crystals in soft tissues can be classified into three main categories: (1) calcinosis, (2) dystrophic calcification, and (3) metastatic calcification. In the presence of normal homeostasis of phosphate, calcinosis and/or dystrophic calcification occur, most often in subcutaneous tissues, skin, and related connective tissues, whereas, in the second case, calcification occurs in degenerated or necrotic tissue. Metastatic calcification occurs in normal tissues when the calcium levels are elevated in serum.
3. Phosphate Homeostasis
In adults, normal phosphate concentration in serum or plasma is mainly 2.5 to 4.5 mg/dL (0.81 to 1.45 mmol/L). Elevated serum phosphate (hyperphosphatemia) is a key risk factor for pathologic calcification in cardiovascular structures [29]. Treatment of hyperphosphatemia with phosphate binders is associated with the slow progression of cardiovascular calcification in hemodialysis patients [30]. Therefore, the homeostasis of phosphate plays a critical role in the initiation and progression of calcification [31] (see Figure 2).
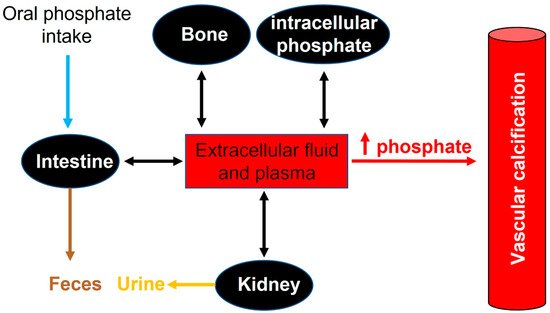
Figure 2. Phosphate flux between body compartments. Phosphate balance is a complex process involving bone intestinal absorption and dietary phosphate and renal excretion of phosphate.
Many different foods contain phosphorus, including vegetables, grains, legumes, eggs, fish, and meats. In addition, phosphate additives such as phosphoric acid, sodium phosphate, and sodium polyphosphate are present in many processed food products. Phosphate deficiency is rare in Europe, and is rarely the result of low dietary intakes. However, phosphate is also available in dietary supplements containing only phosphate o supplements containing phosphate in combination with other ingredients, including vitamins and minerals.
Recommended dietary daily phosphorus intake in healthy adults (>18 years old) is 700 mg. However, daily phosphorus intake varies between 700 and 2000 mg. After absorption, phosphorus is transported across cell membranes as phosphate. And in extracellular fluid (including plasma), phosphate undergoes one of three fates: (1) elimination, mainly by the kidneys, (2) transport into cells, or (3) deposition in bone or soft tissue (see Figure 2).
In healthy adults, oral phosphate intake is balanced mainly by phosphate excretion in the urine and feces. In this case, different factors play an important role in the control of phosphate homeostasis, including phosphate excretion and absorption by the kidneys, intestines, and bone. Although the kidney is the major regulator of phosphate homeostasis, plasma phosphate levels are altered by intestinal phosphate absorption. Notably, in normal adults, between 75% and 85% of the daily phosphate filtered by the glomerulus is reabsorbed by the renal tubules (mainly the proximal tubule) [32][33].
An increased absorption or decreased phosphate excretion can induce a relatively small elevation in serum phosphate, which has been correlated with the presence of calcified vessels due to an increase in calcium-phosphate crystal formation and saturation in the inhibition. Several diseases have been correlated with the dysregulation of phosphate homeostasis, including osteoporosis, diabetes mellitus, hyperparathyroidism, vitamin D (hyper-and hypovitaminosis), and chronic renal disease [34].
4. Phosphate Transporters
Cellular phosphate levels are controlled by sodium-phosphate co-transporters (NaPi) [16][35]. The roles of sodium-phosphate cotransporters in human clinical disease and physiology processes have not been yet well defined. Two families of sodium-phosphate cotransporters have been principally identified, each with multiple members: Type II (also called SLC34 or NaPi-II) and type III (SLC20 or NaPi-III), which transport phosphate with high affinity (Km ≈ 0, 1 or less) but show differences in their affinities for H2PO4− and HPO42− ions [36][37]. Originally identified as a phosphate transporter, Type I (SLC17 or NaPi-I) phosphate transporters mediate the transmembrane transport of organic anions, with relativity low affinity for phosphate suggesting that they transported organic and inorganic anions more readily than phosphate [38].
The SCL34 family comprises three members (also called NaPi-II), which are expressed in the small intestine (NaPi-IIb) and the kidney (NaPi-IIa and NaPi-IIc), two important sites for the control of phosphate homeostasis [39]. NaPi-IIa is expressed predominantly in renal proximal tubules, and under normal conditions, NaPiIIa is the transporter responsible for 95% of phosphate reabsorption in the proximal tubule. An expression of NaPi-IIc was found exclusively in the kidney and described as being growth-related [40][41]. Moreover, the SLC20 family of solute carriers are represented by Pit-1 and Pit-2 (Type III sodium-phosphate cotransporters) [42]. Both cotransporters mediate the movement of phosphate ions across the cell membrane and are ubiquitously expressed, suggesting a “housekeeping” function. More precise localization studies revealed different levels of Pit-1 and Pit-2 expression in each cell type [3].
The roles of sodium phosphate cotransporters in pathophysiology have not been well defined, but their important role in controlling phosphate homeostasis and intracellular phosphate levels for the synthesis of macromolecules and energy metabolism make them an important target to study.
This entry is adapted from the peer-reviewed paper 10.3390/ijms222413536
References
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 2000, 87, E10–E17.
- Li, X.; Yang, H.-Y.; Giachelli, C.M. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ. Res. 2006, 98, 905–912.
- Villa-Bellosta, R.; Bogaert, Y.E.; Levi, M.; Sorribas, V. Characterization of phosphate transport in rat vascular smooth muscle cells: Implications for vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1030–1036.
- Villa-Bellosta, R.; Sorribas, V. Calcium phosphate deposition with normal phosphate concentration. -Role of pyrophosphate-. Circ. J. 2011, 75, 2705–2710.
- Villa-Bellosta, R. Synthesis of Extracellular Pyrophosphate Increases in Vascular Smooth Muscle Cells During Phosphate-Induced Calcification. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2137–2147.
- Schinke, T.; Karsenty, G. Vascular calcification—A passive process in need of inhibitors. Nephrol. Dial. Transplant. 2000, 15, 1272–1274.
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis regulates human vascular calcification in vitro: Evidence for initiation of vascular calcification by apoptotic bodies. Circ. Res. 2000, 87, 1055–1062.
- Shroff, R.C.; McNair, R.; Figg, N.; Skepper, J.N.; Schurgers, L.; Gupta, A.; Hiorns, M.; Donald, A.E.; Deanfield, J.; Rees, L.; et al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation 2008, 118, 1748–1757.
- Mansfield, K.; Teixeira, C.C.; Adams, C.S.; Shapiro, I.M. Phosphate ions mediate chondrocyte apoptosis through a plasma membrane transporter mechanism. Bone 2001, 28, 1–8.
- Steitz, S.A.; Speer, M.Y.; Curinga, G.; Yang, H.Y.; Haynes, P.; Aebersold, R.; Schinke, T.; Karsenty, G.; Giachelli, C.M. Smooth muscle cell phenotypic transition associated with calcification: Upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ. Res. 2001, 89, 1147–1154.
- Li, X.; Yang, H.-Y.; Giachelli, C.M. BMP-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis 2008, 199, 271–277.
- Speer, M.Y.; Li, X.; Hiremath, P.G.; Giachelli, C.M. Runx2/Cbfa1, but not loss of myocardin, is required for smooth muscle cell lineage reprogramming toward osteochondrogenesis. J. Cell. Biochem. 2010, 110, 935–947.
- Villa-Bellosta, R.; Millan, A.; Sorribas, V. Role of calcium-phosphate deposition in vascular smooth muscle cell calcification. Am. J. Physiol. Cell Physiol. 2011, 300, C210–C220.
- Lei, Y.; Sinha, A.; Nosoudi, N.; Grover, A.; Vyavahare, N. Hydroxyapatite and calcified elastin induce osteoblast-like differentiation in rat aortic smooth muscle cells. Exp. Cell Res. 2014, 323, 198–208.
- Sage, A.P.; Lu, J.; Tintut, Y.; Demer, L.L. Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int. 2011, 79, 414–422.
- Villa-Bellosta, R. Vascular Calcification Revisited: A New Perspective for Phosphate Transport. Curr. Cardiol. Rev. 2015, 11, 341–351.
- Kanzaki, N.; Treboux, G.; Onuma, K.; Tsutsumi, S.; Ito, A. Calcium phosphate clusters. Biomaterials 2001, 22, 2921–2929.
- Johnsson, M.S.; Nancollas, G.H. The role of brushite and octacalcium phosphate in apatite formation. Crit. Rev. Oral Biol. Med. 1992, 3, 61–82.
- P’ng, C.H.; Boadle, R.; Horton, M.; Bilous, M.; Bonar, F. Magnesium whitlockite of the aorta. Pathology 2008, 40, 539–540.
- Reid, J.D.; Andersen, M.E. Medial calcification (whitlockite) in the aorta. Atherosclerosis 1993, 101, 213–224.
- Kay, M.I.; Young, R.A.; Posner, A.S. Crystal structure of hydroxyapatite. Nature 1964, 204, 1050–1052.
- Posner, A.S. The structure of bone apatite surfaces. J. Biomed. Mater. Res. 1985, 19, 241–250.
- Posner, A.S.; Beebe, R.A. The surface chemistry of bone mineral and related calcium phosphates. Semin. Arthritis Rheum. 1975, 4, 267–291.
- Posner, A.S.; Betts, F.; Blumenthal, N.C. Role of ATP and Mg in the stabilization of biological and synthetic amorphous calcium phosphates. Calcif. Tissue Res. 1977, 22, 208–212.
- Blumenthal, N.C.; Betts, F.; Posner, A.S. Stabilization of amorphous calcium phosphate by Mg and ATP. Calcif. Tissue Res. 1977, 23, 245–250.
- Urry, D.W. Neutral sites for calcium ion binding to elastin and collagen: A charge neutralization theory for calcification and its relationship to atherosclerosis. Proc. Natl. Acad. Sci. USA 1971, 68, 810–814.
- Khavandgar, Z.; Roman, H.; Li, J.; Lee, S.; Vali, H.; Brinckmann, J.; Davis, E.C.; Murshed, M. Elastin haploinsufficiency impedes the progression of arterial calcification in MGP-deficient mice. J. Bone Miner. Res. 2014, 29, 327–337.
- Hosaka, N.; Mizobuchi, M.; Ogata, H.; Kumata, C.; Kondo, F.; Koiwa, F.; Kinugasa, E.; Akizawa, T. Elastin degradation accelerates phosphate-induced mineralization of vascular smooth muscle cells. Calcif. Tissue Int. 2009, 85, 523–529.
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711.
- Azpiazu, D.; González-Parra, E.; Egido, J.; Villa-Bellosta, R. Hydrolysis of Extracellular Pyrophosphate increases in post-hemodialysis plasma. Sci. Rep. 2018, 8, 11089.
- Villa-Bellosta, R.; Egido, J. Phosphate, pyrophosphate, and vascular calcification: A question of balance. Eur. Heart J. 2017, 38, 1801–1804.
- Manghat, P.; Sodi, R.; Swaminathan, R. Phosphate homeostasis and disorders. Ann. Clin. Biochem. 2014, 51 Pt 6, 631–656.
- Christov, M.; Jüppner, H. Phosphate homeostasis disorders. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 685–706.
- Bergwitz, C.; Jüppner, H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu. Rev. Med. 2010, 61, 91–104.
- Gonzalo, S.; Villa-Bellosta, R. The role of sodium phosphate cotransporters in ectopic calcification. Endokrynol. Pol. 2019, 70, 496–503.
- Forster, I.C.; Hernando, N.; Biber, J.; Murer, H. Phosphate transporters of the SLC20 and SLC34 families. Mol. Asp. Med. 2013, 34, 386–395.
- Forster, I.C.; Hernando, N.; Biber, J.; Murer, H. Phosphate transport kinetics and structure-function relationships of SLC34 and SLC20 proteins. Curr. Top. Membr. 2012, 70, 313–356.
- Reimer, R.J. SLC17: A functionally diverse family of organic anion transporters. Mol. Asp. Med. 2013, 34, 350–359.
- Wagner, C.A.; Hernando, N.; Forster, I.C.; Biber, J. The SLC34 family of sodium-dependent phosphate transporters. Pflugers Arch. 2014, 466, 139–153.
- Silverstein, D.M.; Barac-Nieto, M.; Murer, H.; Spitzer, A. A putative growth-related renal Na(+)-Pi cotransporter. Am. J. Physiol. 1997, 273 Pt 2, R928–R933.
- Segawa, H.; Kaneko, I.; Takahashi, A.; Kuwahata, M.; Ito, M.; Ohkido, I.; Tatsumi, S.; Miyamoto, K.-I. Growth-related renal type II Na/Pi cotransporter. J. Biol. Chem. 2002, 277, 19665–19672.
- Collins, J.F.; Bai, L.; Ghishan, F.K. The SLC20 family of proteins: Dual functions as sodium-phosphate cotransporters and viral receptors. Pflugers Arch. 2004, 447, 647–652.
This entry is offline, you can click here to edit this entry!