Amyloids are filamentous protein aggregates that are associated with a number of incurable diseases, termed amyloidoses. Amyloids can also manifest as infectious or heritable particles, known as prions. While just one prion is known in humans and animals, more than ten prion amyloids have been discovered in fungi. The propagation of fungal prion amyloids requires the chaperone Hsp104, though in excess it can eliminate some prions. Even though Hsp104 acts to disassemble prion fibrils, at normal levels it fragments them into multiple smaller pieces, which ensures prion propagation and accelerates prion conversion. Animals lack Hsp104, but disaggregation is performed by the same complement of chaperones that assist Hsp104 in yeast—Hsp40, Hsp70, and Hsp110. Exogenous Hsp104 can efficiently cooperate with these chaperones in animals and promotes disaggregation, especially of large amyloid aggregates, which indicates its potential as a treatment for amyloid diseases. However, despite the significant effects, Hsp104 and its potentiated variants may be insufficient to fully dissolve amyloid.
- amyloid
- prion
- chaperone
- amyloid fragmentation
- Sup35
- α-synuclein
- Hsp40
- Hsp70
- Hsp104
- HSP110
1. Introduction
2. The Sup35 Protein and Its Prion Structures
2.1. Sup35 Protein Function and Architecture
2.2. [PSI+] Prion Variants
2.3. Sup35 Prion Structures
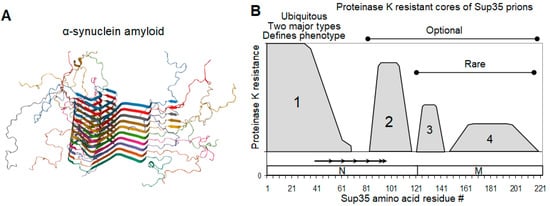
2.4. On the Equivalence of Sup35 In Vitro Fibrils and In Vivo Prions
3. Mechanisms for the Fragmentation and Disassembly of Amyloids in Yeast and Animals
3.1. Replication of Yeast Prions
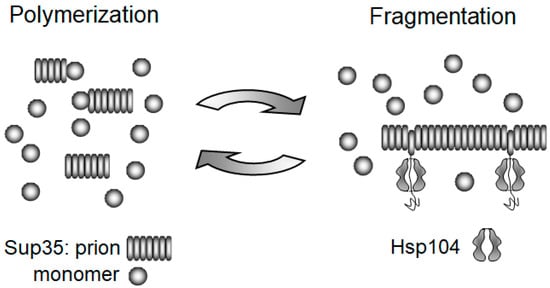
3.2. The Chaperone-Mediated Fragmentation of Yeast Prions
3.3. Amyloid Fragmentation in Animals
3.4. Yeast Prions Based on Putative “Soft” Amyloids or Non-Amyloid Structures
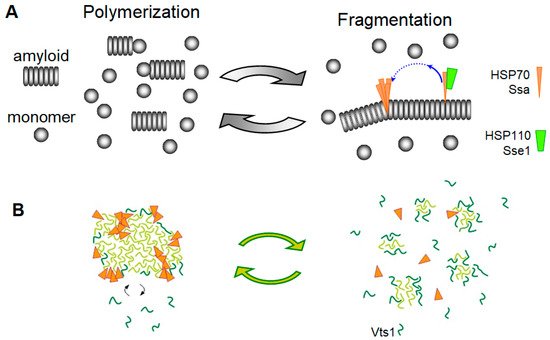
- Elimination of Prions and Amyloids by Hsp104 and Related Chaperones
4.1. Overproduced Hsp104 Acts Differently at the Two Levels of Yeast Prion Structure
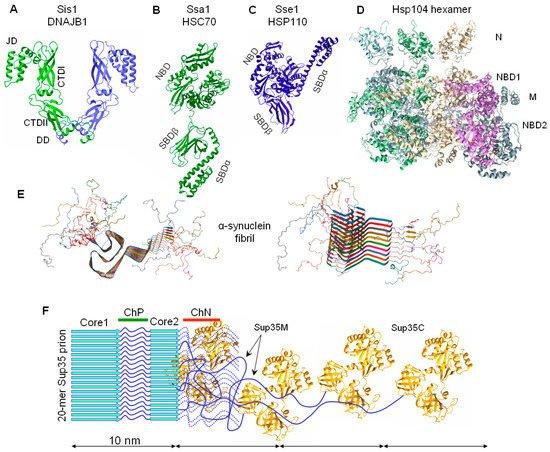
Regrettably, many studies of yeast prions do not differentiate between prion fibrils, prion aggregates, and propagons. We observed that the Sup35 prion particles represent higher order aggregates composed of a number of relatively short fibrils made up of 10–50 Sup35 protomers, as well as some additional proteins [44]. A propagon is a genetic entity defined by the assay developed by Cox et al. [104] where the multiplication of prion particles is supposed to be blocked by GuHCl and these particles are allowed to segregate to individual cells, while an original cell divides for several generations and forms a small colony. This colony is then spread to single cells on a plate that lacks GuHCl and the propagons are then counted as the number of colonies retaining prions. Thus, propagons should be equivalent to prion higher order aggregates, provided that these aggregates neither merge nor split during their growth in the presence of GuHCl. GuHCl fully blocks the fragmentation of prion fibrils [44], but the same was not strictly shown for aggregates. Propagons are clearly different from prion fibrils. Strong [PSI+] fibrils include, on average, about 20 protomers [44]. A yeast cell with strong [PSI+] variants contains about 80,000 Sup35 molecules [105], 4000 fibrils, and 200–1000 propagons [65,106]. Thus, each propagon from a strong [PSI+] contains, on average, 4 to 20 Sup35 fibrils. While fibrils have a strong amyloid structure that is insoluble in SDS, higher order aggregation is based on some weaker interactions that are sensitive to SDS [44].
Our old experiments revealed paradoxical details regarding the effect of overproduced Hsp104 (multicopy HSP104 on a glucose medium) on Sup35 prion aggregates. The size of prion aggregates (as revealed by centrifugation) substantially decreased, while the size of prion fibrils (as assayed by agarose gel electrophoresis) increased or remained constant (Figure 5). These effects were observed for strong [PSI+] variants and artificial prions with the Sup35 N domain from the yeast Pichia methanolica [44,107], while weak [PSI+] variants could not propagate in such conditions [45]. The differential fate of aggregates and fibrils was not understood previously, but it can be explained now with the aid of later observations. We observed that the Sup35 N and M domains in their prion state contained both amyloid structured and unstructured regions [45]. The latter are likely to mediate the aggregation of prion fibers either directly or through chaperones, since the aggregation is sensitive to Hsp104. The fibril-fragmenting activity of Hsp104 is likely to be restricted not by Hsp104 itself, but by the limited amounts of Sis1 and Ssa that are able to bind to a fibril due to the very close spacing of protomers in amyloids (Figure 3). The proportion of Ssa1 and Ssa2 proteins to Sup35 observed in ex vivo [PSI+] prions is about 1:2 [72], which is similar to the maximal value observed for α-syn amyloids [94]. Thus, it appears that Sup35 prion fibrils are already saturated with Ssa, since they have the same 0.48 nm protomer spacing as α-syn amyloids. The Hsp104 fragmenting activity cannot be increased since it depends on the amount of prion-bound Ssa, which also cannot be increased. This could explain why the size of the Sup35 fibrils, in many cases, does not change in response to the strong overproduction of Hsp104. The interactions observed between fibrils in the higher-order aggregates do not have such restrictive architecture and, thus, can be responsive to an increase in Hsp104.
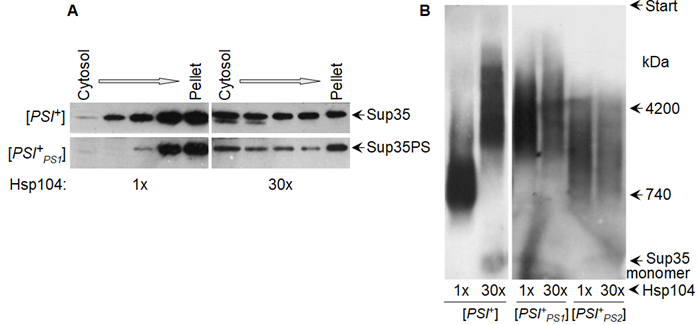
Figure 5. Overproduced Hsp104 disassembles Sup35 prion aggregates, but not fibrils. Hsp104 was either endogenous, or produced from a multicopy plasmid under control of the native promoter (~30-fold excess). (A) Sup35 aggregates of yeast lysates fractionated by centrifugation. Sup35PS, [PSI+PS1], and [PSI+PS2] relate to Sup35 with prion domain from yeast P. methanolica and M and С domains from S. cerevisiae and its two prion variants. (B) SDD-AGE of Sup35 prion fibrils. (A,B) Western blot staining for Sup35NM. Data reproduced from [107] (A) and [44] (B).
4.2. Elimination of Yeast Prions by Hsp104
Regarding the attempts to disassemble amyloids in animal models with the help of Hsp104 that is described in the last chapter, it is of interest to consider the Hsp104-mediated curing of prions in yeast. The curing data are paradoxical. Endogenous Hsp104 can cure a high proportion of [PSI+] variants that emerge in the presence of the weakened Hsp104 mutant, T160M [42,108], and mildly overproduced Hsp104 can cure some [PSI+] variants [58], but some other variants are not cured even at high Hsp104 overproduction [45].
The data on prion curing by strongly overproduced Hsp104 are contradictory. Though initially it appeared that Hsp104 overproduction should proportionally increase its prion fragmenting and disaggregating activities [29], this is apparently not so. It is often stated that [PSI+] may be the only yeast prion that can be efficiently cured by excess Hsp104, since [URE3] and [PIN+] are resistant to such impacts [109,110]. However, some level of [URE3] curing has been shown in a later work [111]. Among [PSI+] variants, weak ones are readily cured by excess Hsp104, while strong [PSI+] variants are more resistant. This difference depends somewhat on the specifics of Hsp104 overproduction. When the HSP104 gene was expressed from a multicopy 2-micron plasmid (this results in about 30-fold Hsp104 overproduction [112]), in the testing of eight weak and seven strong [PSI+] isolates, all the weak ones were fully cured, while all strong ones were fully resistant [45]. Production under a low copy inducible GAL1 promoter results in a similar 20–40 fold Hsp104 excess, but it cures a strong [PSI+] variant almost completely in ten generations of growth [106]. The difference in the curing of strong [PSI+] variants appears to depend on the carbon source (galactose versus glucose) [45]. We also observed (unpublished data) that the absence or the malfunction of mitochondria allows the complete curing of strong [PSI+] variants by the multicopy of HSP104 on glucose.
The easier curing of weak Sup35 prions appears paradoxical, since they should be less efficiently recognized and disassembled by chaperones compared to the strong ones, as suggested by their larger fibrils [44] and higher mechanical strength [56,89]. Greene et al. proposed that the curing of Sup35 prions mainly occurs through the Hsp104-mediated trimming of prion fibers from the ends. The extraction of Sup35 protomers from the prion ends does not generate new prion particles and it is likely to proceed relatively fast, since the ends of a fiber could be more accessible and the terminal protomers are easier to unfold as they are joined to just one other protomer, rather than two. These authors tried to confirm this idea by microscopic observation of the dissolution of the aggregates of GFP-labeled prion particles, after the start of Hsp104 overproduction, in several reviewed works [113]. However, this approach does not appear adequate for the question. The visible prion–GFP foci represent large aggregates, including many prion fibrils [44]. As we already noted, overproduced Hsp104 can disassemble these aggregates into smaller particles, down to single fibrils [107] that are undistinguishable from monomers by light microscopy, without decreasing the size of these fibrils [44]). The best way to observe trimming would be to monitor the size of prion fibrils by agarose electrophoresis, called SDD-AGE [44], but this was not done.
Serio et al. proposed that Sup35 prion fibrils smaller than a certain threshold size may be unstable and would be dissolved, and that such a threshold is higher for weak prions. Hsp104 action not just at, but also near, prion fiber ends would act as a trimming process, generating no new prion particles; the higher the minimal size of weak Sup35 prions could explain their easier curing by Hsp104. However, while a mathematical model of this effect was provided, the experimental evidence in support of this idea [114] does not appear sufficient. It is also unclear why weak Sup35 prions, which are mechanically sturdier than strong prions [56] and less well-recognized by chaperones, require more protomers to stabilize their minimal particles.
A more convincing group of data, although still partly contradictory, relates the [PSI+] curing by overproduced Hsp104 to prion malpartition, rather than dissolution. The initial observation was that Hsp104 can bind directly to the Sup35 M domain region 129–148, which is important for curing, since Sup35 prions lacking this region are not cured by Hsp104 overproduction [33].This Hsp104 binding is independent of Hsp40 and Hsp70 and, in relation to this, nonproductive [66]. In line with this, in vivo Hsp104 shows two types of binding to Sup35 prions: one is labile, Hsp70-dependent, and exhibits a free exchange of Hsp104 with the pool of monomers; the other is a stable binding to the Sup35 M region that shows little exchange [115]. The latter could be termed a direct and non-productive binding. Such a binding should sterically interfere or compete with the binding of different chaperones belonging to the productive fragmentation pathway, due to the relatively small space where such interactions occur (Figure 3), and this could thus reduce prion fragmentation. In agreement with this, overproduced Hsp104 can substantially increase the size of strong Sup35 prion fibrils (Figure 5B) [44]. In contrast, Ness et al observed that Hsp104 overproduction under a low copy GAL1 promoter did not impair fragmentation and did not alter Sup35 oligomer size. In their setup, the curing of strong [PSI+] variants, which occurred at a rate of about 10% per generation, was shown to be due to prion retention in a proportion of mother cells. These authors proposed that Hsp104 mediated prion binding to some subcellular structures, thus causing their malpartition [106]. Regrettably, such a study was not made for weak [PSI+] variants where curing would be more pronounced.
[PSI+] variants can also be cured with moderate efficiency by short-term heat shock. Such a shock increases the proportion of Hsp104 to Ssa proteins, and the prion curing also occurs through the asymmetric segregation of the prion [116,117].
Notably, the weak [PSI+] variant shows a larger proportion of stable Hsp104 binding to the Sup35 M domain [115], which can explain the easier curing of weak [PSI+] variants. In turn, the more efficient direct binding to weak Sup35 prions could be explained by our recent data. The Hsp104 target region of Sup35 is 129-148, which coincides well with amyloid Core 3 (124–153) that forms in the majority of strong [PSI+] variants but is rarely found in weak [PSI+] variants [45]. It is reasonable to assume that Hsp104 can bind the target region in its unfolded state, but not while in an amyloid fold, and so non-productive Hsp104 binding is less likely in strong [PSI+] variants.
On the other hand, the productive binding of Hsp104 through Sis1 and Ssa should be more efficient in strong [PSI+] variants. These chaperones bind to the unfolded regions of the Sup35 N domain, rich in tyrosine, which stimulates fragmentation best [57,118]. Such regions are smaller in weak Sup35 prions due to the presence of Core 2 (~90–123) amyloid structure that is rare, or less pronounced, in strong [PSI+] variants [45]. Thus, the reduced productive and increased non-productive Hsp104 binding could define the easier curing of weak [PSI+] variants by overproduced Hsp104.
Thus, despite some experimental discrepancies, the only confirmed mechanism of [PSI+] curing by overproduced Hsp104 is the inefficient partitioning of propagons in cell divisions, due to either reduced prion fragmentation, or Hsp104-mediated prion anchoring. Prion curing through its dissolution currently lacks sufficient evidence. Curing by normal Hsp104 levels of the prions generated in the presence of mutant Hsp104 is of great interest in this respect, but its mechanism has not been studied in detail.
4.3. The Therapeutic Potential of Protein Disaggregases
While the human Hsc70 system can efficiently disaggregate toxic oligomers and short amyloid fibrils, its activity against large, less toxic amyloid aggregates is severely impaired [119]. Yeast Hsp104 is a much more powerful disaggregase than the Hsp70 (Ssa)-Hsp40 (Sis1) system alone [93], but animals have no Hsp104 homolog [90]. This raised hope that Hsp104 can disaggregate pathological amyloids if reintroduced to animal cells [120,121]. Luckily, Hsp104 can efficiently collaborate with the animal disaggregation machinery and strongly improves the reactivation of heat-denatured luciferase [93,122]. In vitro, Hsp104 can dissolve fibrils associated with human diseases: amyloid β, α-syn, prion protein, tau, amylin, and polyglutamine [89,122,123]. In animal disease models, Hsp104 reduces polyglutamine toxicity in Caenorhabditis elegans, fly, and rodent models [124–127]. In a rat model of Parkinson’s disease, Hsp104 reduced the formation of phosphorylated α-syn inclusions and prevented nigrostriatal dopaminergic neurodegeneration [122].
However, the activity of yeast Hsp104 against pathological aggregates can be insufficient even at high levels of Hsp104 [89]. This prompted attempts to enhance Hsp104 disaggregation activities, and this was, surprisingly, achieved through minor changes and even single missense mutations [128]. Potentiated Hsp104 variants have been developed that are capable of suppressing toxicity associated with α-syn, TDP-43, and FUS in yeast [129–133]. Many such potentiating mutations were found in the regulatory coiled-coil middle (M) domain of Hsp104 (residues 411–538), which mediates interactions of Hsp104 with Hsp70. Hsp104 potentiation often correlates with the destabilization of the M domain. However, some of these mutations show off-target toxicity. To overcome this problem, scanning mutagenesis of the M domain was performed [134], as well as mutagenesis of Hsp104 NBD1 and NBD2 [129,133], which allowed the isolation of non-toxic potentiated Hsp104 mutants. The screening of a cross-kingdom collection of Hsp104 homologs in yeast proteotoxicity models revealed that prokaryotic ClpG reduces TDP-43, FUS, and α-syn toxicity, whereas prokaryotic ClpB is ineffective. The latter is not surprising, since Reidy et al. showed that bacterial ClpB did not properly interact with yeast chaperones and required its bacterial partner chaperones to function [135]. Distinct eukaryotic Hsp104 homologs were uncovered that selectively antagonized α-syn condensation and toxicity in yeast and dopaminergic neurodegeneration in C. elegans. Surprisingly, this therapeutic variation does not manifest as enhanced disaggregase activity, but rather as an increased passive inhibition of the aggregation of specific substrates [136]. An Hsp104 variant that is efficient against TDP-43, α-syn, and polyglutamine, that lacked toxicity, was obtained from the thermophilic fungus Calcarisporiella thermophila [137].
While animals lack the mitochondrial Hsp104 homolog, Hsp78, disaggregation in mitochondria can be performed by Skd3, another chaperone of the AAA+ family. Skd3 shows a homology with bacterial ClpB, but has only one NBD. Mutations in Skd3 that reduce its disaggregating activities are associated with 3-methylglutaconic aciduria, a severe mitochondrial disorder. Thus, Skd3 is a potent mitochondrial protein disaggregase which can be used for treating mitochondrial protein aggregation [138].
AAA+ proteins that do not belong to the Hsp104 family can also be used to counteract toxic protein misfolding in animals. One such protein is archaeal PAN, an unfoldase homologous to the eukaryotic proteosomal 19S particle. PAN associates with the 20S catalytic particle and unfolds substrates before their degradation [139]. A PAN variant was recently constructed with a C-terminal FLAG epitope tag (PANet), which impedes PAN interactions with the 20S proteasome, but does not affect unfolding. The expression of PANet in rod photoreceptors in a mouse model of retinopathy mitigates photoreceptor degeneration caused by protein misfolding without causing significant side effects [140]. Thus, protein disaggregases of the AAA+ family have significant therapeutic potential.
It is important to keep in mind that the disaggregation of amyloids can have both positive and negative effects. Negative consequences might occur when large amyloids are broken into smaller pieces, but the latter are not efficiently destroyed. This resembles the situation with yeast prions, which are propagated by Hsp104. In animals, amyloids of smaller size are (1) often more toxic and (2) have a much higher potential for a prion-like spread between cells and tissues. Such problems were highlighted recently by Tittelmeier et al. who observed that reducing disaggregation in C. elegans by knocking down Hsp110 caused a beneficial decrease of the amounts of toxic α-syn species and a reduction in the intercellular propagation of α-syn aggregates. A similar treatment decreased the amount of polyQ aggregates and their toxic effects in another C. elegans model [141]. This suggests that the optimal disaggregation activity would be one that destroys small aggregates, but cannot cope with large ones. Humans have three NEFs for Hsp70, which stimulate the entropic pulling effect to a different extent [94]. Possibly, optimal activity might be achieved by adjusting levels of these NEFs.
The described data raises certain hopes and reveals some fundamental problems. The most optimistic aim would be the complete dissolution of amyloids, but this has never been shown, even in yeast. Yeast prions can be cured through retention in the mother cell, but such a scenario is not relevant for multicellular organisms, where most cells do not actively divide. Still, some hope comes from observations in yeast where prions that appear in the presence of weakened Hsp104 are less resistant to Hsp104, while human amyloids appear in the absence of Hsp104. However, they possess multiple mechanisms for the intercellular movement of amyloids (reviewed in previous research [142]) and these are likely to be affected by factors, which influence prion retention in dividing yeast cells.
A central technical problem would be to deliver Hsp104 to every cell or extracellular location containing an amyloid, which does not seem currently feasible. Finally, a tool able to disassemble all amyloids could also disassemble functional amyloids, and, in particular, amyloids that are involved in long-term memory.
Nevertheless, disaggregases of the Hsp104 type can alleviate the symptoms of amyloidoses in some animal models. Although there might not be a single ideal disaggregase, different agents tailored for each type of amyloidosis might be possible. In any case, the most promising strategy would be to adjust disaggregases so that they disassembled more toxic, but less resistant, small amyloids, while leaving less toxic and more resistant larger aggregates intact.
References
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress over the Last Decade. Rev. Biochem. 2017, 86, 27–68, doi:10.1146/annurev-biochem-061516-045115.
- Prusiner, S.B. Biology and genetics of prions causing neurodegeneration. Rev. Genet. 2013, 47, 601–623, doi:10.1146/annurev-genet-110711-155524.
- Brundin, P.; Melki, R.; Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Rev. Mol. Cell Biol. 2010, 11, 301–307, doi:10.1038/nrm2873.
- Goedert, M.; Masuda-Suzukake, M.; Falcon, B. Like prions: The propagation of aggregated tau and α-synuclein in neurodegeneration. Brain 2017, 140, 266–278, doi:10.1093/brain/aww230.
- Lam, S.; Petit, F.; Hérard, A.-S.; Boluda, S.; Eddarkaoui, S.; Guillermier, M.; Letournel, F.; Martin-Négrier, M.-L.; Faisant, M.; Godfraind, C.; et al. Transmission of amyloid-beta and tau pathologies is associated with cognitive impairments in a primate. Acta Neuropathol. Commun. 2021, 9, 165, doi:10.1186/s40478-021-01266-8.
- Keleman, K.; Krüttner, S.; Alenius, M.; Dickson, B.J. Function of the Drosophila CPEB protein Orb2 in long-term courtship memory. Neurosci. 2007, 10, 1587–1593, doi:10.1038/nn1996.
- Mastushita-Sakai, T.; White-Grindley, E.; Samuelson, J.; Seidel, C.; Si, K. Drosophila Orb2 targets genes involved in neuronal growth, synapse formation, and protein turnover. Natl. Acad. Sci. USA 2010, 107, 11987–11992, doi:10.1073/pnas.1004433107.
- Wickner, R.B. Yeast and Fungal Prions. Cold Spring Harb. Perspect. Biol. 2016, 8, a023531, doi:10.1101/cshperspect.a023531.
- Cox, B.S. Ψ, A cytoplasmic suppressor of super-suppressor in yeast. Heredity (Edinb) 1965, 20, 505–521.
- Cox, B.; Tuite, M. The life of [PSI]. Genet. 2018, 64, 1–8, doi:10.1007/s00294-017-0714-7.
- Lacroute, F. Non-Mendelian mutation allowing ureidosuccinic acid uptake in yeast. Bacteriol. 1971, 106, 519–522, doi:10.1128/jb.106.2.519-522.1971.
- Wickner, R.B. [URE3] as an altered URE2 protein: Evidence for a prion analog in Saccharomyces cerevisiae. Science 1994, 264, 566–569, doi:10.1126/science.7909170.
- Derkatch, I.L.; Bradley, M.E.; Hong, J.Y.; Liebman, S.W. Prions affect the appearance of other prions: The story of [PIN+]. Cell 2001, 106, 171–182, doi:10.1016/s0092-8674(01)00427-5.
- Du, Z.; Park, K.-W.; Yu, H.; Fan, Q.; Li, L. Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Genet. 2008, 40, 460–465, doi:10.1038/ng.112.
- Patel, B.K.; Gavin-Smyth, J.; Liebman, S.W. The yeast global transcriptional co-repressor protein Cyc8 can propagate as a prion. Cell Biol. 2009, 11, 344–349, doi:10.1038/ncb1843.
- Suzuki, G.; Shimazu, N.; Tanaka, M. A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress. Science 2012, 336, 355–359, doi:10.1126/science.1219491.
- Alberti, S.; Halfmann, R.; King, O.; Kapila, A.; Lindquist, S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009, 137, 146–158, doi:10.1016/j.cell.2009.02.044.
- Halfmann, R.; Wright, J.R.; Alberti, S.; Lindquist, S.; Rexach, M. Prion formation by a yeast GLFG nucleoporin. Prion 2012, 6, 391–399, doi:10.4161/pri.20199.
- Chernova, T.A.; Kiktev, D.A.; Romanyuk, A.V.; Shanks, J.R.; Laur, O.; Ali, M.; Ghosh, A.; Kim, D.; Yang, Z.; Mang, M.; et al. Yeast Short-Lived Actin-Associated Protein Forms a Metastable Prion in Response to Thermal Stress. Cell Rep. 2017, 18, 751–761, doi:10.1016/j.celrep.2016.12.082.
- Cereghetti, G.; Wilson-Zbinden, C.; Kissling, V.M.; Diether, M.; Arm, A.; Yoo, H.; Piazza, I.; Saad, S.; Picotti, P.; Drummond, D.A.; et al. Reversible amyloids of pyruvate kinase couple cell metabolism and stress granule disassembly. Cell Biol. 2021, 23, 1085–1094, doi:10.1038/s41556-021-00760-4.
- Reichert, P.; Caudron, F. Mnemons and the memorization of past signaling events. Opin. Cell Biol. 2021, 69, 127–135, doi:10.1016/j.ceb.2021.01.005.
- Brown, J.C.S.; Lindquist, S. A heritable switch in carbon source utilization driven by an unusual yeast prion. Genes Dev. 2009, 23, 2320–2332, doi:10.1101/gad.1839109.
- Chakrabortee, S.; Byers, J.S.; Jones, S.; Garcia, D.M.; Bhullar, B.; Chang, A.; She, R.; Lee, L.; Fremin, B.; Lindquist, S.; et al. Intrinsically Disordered Proteins Drive Emergence and Inheritance of Biological Traits. Cell 2016, 167, 369–381.e12, doi:10.1016/j.cell.2016.09.017.
- True, H.L.; Lindquist, S.L. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 2000, 407, 477–483, doi:10.1038/35035005.
- Namy, O.; Galopier, A.; Martini, C.; Matsufuji, S.; Fabret, C.; Rousset, J.-P. Epigenetic control of polyamines by the prion [PSI+]. Cell Biol. 2008, 10, 1069–1075, doi:10.1038/ncb1766.
- Tuite, M.F. Yeast models of neurodegenerative diseases. Mol. Biol. Transl. Sci. 2019, 168, 351–379, doi:10.1016/bs.pmbts.2019.07.001.
- Chernoff, Y.O.; Grizel, A.V.; Rubel, A.A.; Zelinsky, A.A.; Chandramowlishwaran, P.; Chernova, T.A. Application of yeast to studying amyloid and prion diseases. Genet. 2020, 105, 293–380, doi:10.1016/bs.adgen.2020.01.002.
- Serpionov, G.V.; Alexandrov, A.I.; Antonenko, Y.N.; Ter-Avanesyan, M.D. A protein polymerization cascade mediates toxicity of non-pathological human huntingtin in yeast. Rep. 2015, 5, 18407, doi:10.1038/srep18407.
- Kushnirov, V.V.; Ter-Avanesyan, M.D. Structure and replication of yeast prions. Cell 1998, 94, 13–16.
- Stansfield, I.; Jones, K.M.; Kushnirov, V.V.; Dagkesamanskaya, A.R.; Poznyakovski, A.I.; Paushkin, S.V.; Nierras, C.R.; Cox, B.S.; Ter-Avanesyan, M.D.; Tuite, M.F. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J. 1995, 14, 4365–4373.
- Zhouravleva, G.; Frolova, L.; Le Goff, X.; Le Guellec, R.; Inge-Vechtomov, S.; Kisselev, L.; Philippe, M. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J. 1995, 14, 4065–4072.
- Ter-Avanesyan, M.D.; Kushnirov, V.V.; Dagkesamanskaya, A.R.; Didichenko, S.A.; Chernoff, Y.O.; Inge-Vechtomov, S.G.; Smirnov, V.N. Deletion analysis of the SUP35 gene of the yeast Saccharomyces cerevisiae reveals two non-overlapping functional regions in the encoded protein. Microbiol. 1993, 7, 683–692, doi:10.1111/j.1365-2958.1993.tb01159.x.
- Helsen, C.W.; Glover, J.R. Insight into molecular basis of curing of [PSI+] prion by overexpression of 104-kDa heat shock protein Hsp104. Biol. Chem. 2012, 287, 542–556, doi:10.1074/jbc.M111.302869.
- Franzmann, T.M.; Jahnel, M.; Pozniakovsky, A.; Mahamid, J.; Holehouse, A.S.; Nüske, E.; Richter, D.; Baumeister, W.; Grill, S.W.; Pappu, R.V.; et al. Phase separation of a yeast prion protein promotes cellular fitness. Science 2018, 359, 6371, doi:10.1126/science.aao5654.
- Mehra, S.; Gadhe, L.; Bera, R.; Sawner, A.S.; Maji, S.K. Structural and Functional Insights into α-Synuclein Fibril Polymorphism. Biomolecules 2021, 11, 1419, doi:10.3390/biom11101419.
- Carta, M.; Aguzzi, A. Molecular foundations of prion strain diversity. Opin. Neurobiol. 2021, 72, 22–31, doi:10.1016/j.conb.2021.07.010.
- Derkatch, I.L.; Chernoff, Y.O.; Kushnirov, V.V.; Inge-Vechtomov, S.G.; Liebman, S.W. Genesis and variability of [PSI+] prion factors in Saccharomyces cerevisiae. Genetics 1996, 144, 1375–1386, doi:10.1093/genetics/144.4.1375.
- Wickner, R.B.; Son, M.; Edskes, H.K. Prion Variants of Yeast are Numerous, Mutable, and Segregate on Growth, Affecting Prion Pathogenesis, Transmission Barriers, and Sensitivity to Anti-Prion Systems. Viruses 2019, 11, 238, doi:10.3390/v11030238.
- Nizhnikov, A.A.; Ryzhova, T.A.; Volkov, K.V.; Zadorsky, S.P.; Sopova, J.V.; Inge-Vechtomov, S.G.; Galkin, A.P. Interaction of Prions Causes Heritable Traits in Saccharomyces cerevisiae. PLoS Genet. 2016, 12, e1006504, doi:10.1371/journal.pgen.1006504.
- Chang, H.-Y.; Lin, J.-Y.; Lee, H.-C.; Wang, H.-L.; King, C.-Y. Strain-specific sequences required for yeast [PSI+] prion propagation. Natl. Acad. Sci. USA 2008, 105, 13345–13350, doi:10.1073/pnas.0802215105.
- Huang, Y.-W.; King, C.-Y. A complete catalog of wild-type Sup35 prion variants and their protein-only propagation. Genet. 2020, 66, 97–122, doi:10.1007/s00294-019-01003-8.
- Huang, Y.-W.; Kushnirov, V.V.; King, C.-Y. Mutable yeast prion variants are stabilized by a defective Hsp104 chaperone. Microbiol. 2021, 115, 774–788, doi:10.1111/mmi.14643.
- Ohhashi, Y.; Yamaguchi, Y.; Kurahashi, H.; Kamatari, Y.O.; Sugiyama, S.; Uluca, B.; Piechatzek, T.; Komi, Y.; Shida, T.; Müller, H.; et al. Molecular basis for diversification of yeast prion strain conformation. Natl. Acad. Sci. USA 2018, 115, 2389–2394, doi:10.1073/pnas.1715483115.
- Kryndushkin, D.S.; Alexandrov, I.M.; Ter-Avanesyan, M.D.; Kushnirov, V. V Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. Biol. Chem. 2003, 278, 49636–49643, doi:10.1074/jbc.M307996200.
- Dergalev, A.; Alexandrov, A.; Ivannikov, R.; Ter-Avanesyan, M.; Kushnirov, V. Yeast Sup35 Prion Structure: Two Types, Four Parts, Many Variants. J. Mol. Sci. 2019, 20, 2633, doi:10.3390/ijms20112633.
- Shewmaker, F.; Wickner, R.B.; Tycko, R. Amyloid of the prion domain of Sup35p has an in-register parallel beta-sheet structure. Natl. Acad. Sci. USA 2006, 103, 19754–19759, doi:10.1073/pnas.0609638103.
- Shewmaker, F.; Kryndushkin, D.; Chen, B.; Tycko, R.; Wickner, R.B. Two prion variants of Sup35p have in-register parallel β-sheet structures, independent of hydration. Biochemistry 2009, 48, 5074–5082, doi:10.1021/bi900345q.
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Struct. Mol. Biol. 2016, 23, 409–415, doi:10.1038/nsmb.3194.
- Krishnan, R.; Lindquist, S.L. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature 2005, 435, 765–772, doi:10.1038/nature03679.
- Toyama, B.H.; Kelly, M.J.S.; Gross, J.D.; Weissman, J.S. The structural basis of yeast prion strain variants. Nature 2007, 449, 233–237, doi:10.1038/nature06108.
- Depace, A.H.; Santoso, A.; Hillner, P.; Weissman, J.S. A Critical Role for Amino-Terminal Glutamine/Asparagine Repeats in the Formation and Propagation of a Yeast Prion. Cell 1998, 93, 1241–1252.
- Huang, Y.W.; Chang, Y.C.; Diaz-Avalos, R.; King, C.Y. W8, a new Sup35 prion strain, transmits distinctive information with a conserved assembly scheme. Prion 2015, 9, 207–227, doi:10.1080/19336896.2015.1039217.
- Ghosh, R.; Dong, J.; Wall, J.; Frederick, K.K. Amyloid fibrils embodying distinctive yeast prion phenotypes exhibit diverse morphologies. FEMS Yeast Res. 2018, 18, foy059, doi:10.1093/femsyr/foy059.
- Parham, S.N.; Resende, C.G.; Tuite, M.F. Oligopeptide repeats in the yeast protein Sup35p stabilize intermolecular prion interactions. EMBO J. 2001, 20, 2111–2119, doi:10.1093/emboj/20.9.2111.
- Shkundina, I.S.; Kushnirov, V.V.; Tuite, M.F.; Ter-Avanesyan, M.D. The role of the N-terminal oligopeptide repeats of the yeast Sup35 prion protein in propagation and transmission of prion variants. Genetics 2006, 172, 827–835, doi:10.1534/genetics.105.048660.
- Tanaka, M.; Collins, S.R.; Toyama, B.H.; Weissman, J.S. The physical basis of how prion conformations determine strain phenotypes. Nature 2006, 442, 585–589, doi:10.1038/nature04922.
- Alexandrov, A.I.; Polyanskaya, A.B.; Serpionov, G.V.; Ter-Avanesyan, M.D.; Kushnirov, V.V. The Effects of Amino Acid Composition of Glutamine-Rich Domains on Amyloid Formation and Fragmentation. PLoS ONE 2012, 7, e46458, doi:10.1371/journal.pone.0046458.
- Chernoff, Y.O.; Lindquist, S.L.; Ono, B.; Inge-Vechtomov, S.G.; Liebman, S.W. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [PSI+]. Science 1995, 268, 880–884, doi:10.1126/science.7754373.
- Paushkin, S.V.; Kushnirov, V.V.; Smirnov, V.N.; Ter-Avanesyan, M.D. Propagation of the yeast prion-like [PSI+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 1996, 15, 3127–3134.
- Parsell, D.A.; Kowal, A.S.; Singer, M.A.; Lindquist, S. Protein disaggregation mediated by heat-shock protein Hsp104. Nature 1994, 372, 475–478, doi:10.1038/372475a0.
- King, C.-Y.; Tittmann, P.; Gross, H.; Gebert, R.; Aebi, M.; Wuthrich, K. Prion-inducing domain 2-114 of yeast Sup35 protein transforms in vitro into amyloid-like filaments. Natl. Acad. Sci. USA 1997, 94, 6618–6622, doi:10.1073/pnas.94.13.6618.
- Glover, J.R.; Kowal, A.S.; Schirmer, E.C.; Patino, M.M.; Liu, J.J.; Lindquist, S. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 1997, 89, 811–819, doi:10.1016/s0092-8674(00)80264-0.
- Patino, M.M.; Liu, J.J.; Glover, J.R.; Lindquist, S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science 1996, 273, 622–626, doi:10.1126/science.273.5275.622.
- Paushkin, S.V.; Kushnirov, V.V.; Smirnov, V.N.; Ter-Avanesyan, M.D. In vitro propagation of the prion-like state of yeast Sup35 protein. Science 1997, 277, 381–383.
- Byrne, L.J.; Cole, D.J.; Cox, B.S.; Ridout, M.S.; Morgan, B.J.T.; Tuite, M.F. The number and transmission of [PSI+] prion seeds (Propagons) in the yeast Saccharomyces cerevisiae. PLoS ONE 2009, 4, e4670, doi:10.1371/journal.pone.0004670.
- Winkler, J.; Tyedmers, J.; Bukau, B.; Mogk, A. Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. Cell Biol. 2012, 198, 387–404, doi:10.1083/jcb.201201074.
- Tuite, M.F.; Mundy, C.R.; Cox, B.S. Agents that cause a high frequency of genetic change from [PSI+] to [psi-] in Saccharomyces cerevisiae. Genetics 1981, 98, 691–711, doi:10.1093/genetics/98.4.691.
- Prusiner, S.B. Prions. Natl. Acad. Sci. USA 1998, 95, 13363–13383, doi:10.1073/pnas.95.23.13363.
- Cohen, F.E.; Pan, K.M.; Huang, Z.; Baldwin, M.; Fletterick, R.J.; Prusiner, S.B. Structural clues to prion replication. Science 1994, 264, 530–531, doi:10.1126/science.7909169.
- Walker, L.C. Proteopathic Strains and the Heterogeneity of Neurodegenerative Diseases. Rev. Genet. 2016, 50, 329–346, doi:10.1146/annurev-genet-120215-034943.
- Glover, J.R.; Lindquist, S. Hsp104, Hsp70, and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell 1998, 94, 73–82.
- Bagriantsev, S.N.; Gracheva, E.O.; Richmond, J.E.; Liebman, S.W. Variant-specific [PSI+] infection is transmitted by Sup35 polymers within [PSI+] aggregates with heterogeneous protein composition. Biol. Cell 2008, 19, 2433–2443, doi:10.1091/mbc.e08-01-0078.
- Park, S.; Wang, X.; Xi, W.; Richardson, R.; Laue, T.M.; Denis, C.L. The non-prion SUP35 preexists in large chaperone-containing molecular complexes. Proteins 2021, doi:10.1002/prot.26282.
- Sondheimer, N.; Lopez, N.; Craig, E.A.; Lindquist, S. The role of Sis1 in the maintenance of the [RNQ+] prion. EMBO J. 2001, 20, 2435–2442, doi:10.1093/emboj/20.10.2435.
- Higurashi, T.; Hines, J.K.; Sahi, C.; Aron, R.; Craig, E.A. Specificity of the J-protein Sis1 in the propagation of 3 yeast prions. Natl. Acad. Sci. USA 2008, 105, 16596–16601, doi:10.1073/pnas.0808934105.
- Tipton, K.A.; Verges, K.J.; Weissman, J.S. In vivo monitoring of the prion replication cycle reveals a critical role for Sis1 in delivering substrates to Hsp104. Cell 2008, 32, 584–591, doi:10.1016/j.molcel.2008.11.003.
- Hines, J.K.; Li, X.; Du, Z.; Higurashi, T.; Li, L.; Craig, E.A. [SWI], the prion formed by the chromatin remodeling factor Swi1, is highly sensitive to alterations in Hsp70 chaperone system activity. PLoS Genet. 2011, 7, e1001309, doi:10.1371/journal.pgen.1001309.
- Schilke, B.A.; Ciesielski, S.J.; Ziegelhoffer, T.; Kamiya, E.; Tonelli, M.; Lee, W.; Cornilescu, G.; Hines, J.K.; Markley, J.L.; Craig, E.A. Broadening the functionality of a J-protein/Hsp70 molecular chaperone system. PLoS Genet. 2017, 13, e1007084, doi:10.1371/journal.pgen.1007084.
- Bradley, M.E.; Edskes, H.K.; Hong, J.Y.; Wickner, R.B.; Liebman, S.W. Interactions among prions and prion “strains” in yeast. Natl. Acad. Sci. USA 2002, 99 (Suppl. 4), 16392–16399, doi:10.1073/pnas.152330699.
- Kryndushkin, D.S.; Smirnov, V.N.; Ter-Avanesyan, M.D.; Kushnirov, V.V. Increased expression of Hsp40 chaperones, transcriptional factors, and ribosomal protein Rpp0 can cure yeast prions. Biol. Chem. 2002, 277, 23702–23708, doi:10.1074/jbc.M111547200.
- Troisi, E.M.; Rockman, M.E.; Nguyen, P.P.; Oliver, E.E.; Hines, J.K. Swa2, the yeast homolog of mammalian auxilin, is specifically required for the propagation of the prion variant [URE3-1]. Microbiol. 2015, 97, 926–941, doi:10.1111/mmi.13076.
- Parsell, D.A.; Kowal, A.S.; Lindquist, S. Saccharomyces cerevisiae Hsp104 protein. Purification and characterization of ATP-induced structural changes. Biol. Chem. 1994, 269, 4480–4487.
- Shorter, J.; Southworth, D.R. Spiraling in Control: Structures and Mechanisms of the Hsp104 Disaggregase. Cold Spring Harb. Perspect. Biol. 2019, 11, a034033, doi:10.1101/cshperspect.a034033.
- Gates, S.N.; Yokom, A.L.; Lin, J.; Jackrel, M.E.; Rizo, A.N.; Kendsersky, N.M.; Buell, C.E.; Sweeny, E.A.; Mack, K.L.; Chuang, E.; et al. Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science 2017, 357, 273–279, doi:10.1126/science.aan1052.
- Avellaneda, M.J.; Franke, K.B.; Sunderlikova, V.; Bukau, B.; Mogk, A.; Tans, S.J. Processive extrusion of polypeptide loops by a Hsp100 disaggregase. Nature 2020, 578, 317–320, doi:10.1038/s41586-020-1964-y.
- Zeymer, C.; Werbeck, N.D.; Schlichting, I.; Reinstein, J. The molecular mechanism of Hsp100 chaperone inhibition by the prion curing agent guanidinium chloride. Biol. Chem. 2013, 288, 7065–7076, doi:10.1074/jbc.M112.432583.
- Kummer, E.; Oguchi, Y.; Seyffer, F.; Bukau, B.; Mogk, A. Mechanism of Hsp104/ClpB inhibition by prion curing Guanidinium hydrochloride. FEBS Lett. 2013, 587, 810–817, doi:10.1016/j.febslet.2013.02.011.
- Escusa-Toret, S.; Vonk, W.I.M.; Frydman, J. Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Cell Biol. 2013, 15, 1231–1243, doi:10.1038/ncb2838.
- Desantis, M.E.; Leung, E.H.; Sweeny, E.A.; Jackrel, M.E.; Cushman-Nick, M.; Neuhaus-Follini, A.; Vashist, S.; Sochor, M.A.; Knight, M.N.; Shorter, J. Operational plasticity enables Hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell 2012, 151, 778–793, doi:10.1016/j.cell.2012.09.038.
- Erives, A.J.; Fassler, J.S. Metabolic and chaperone gene loss marks the origin of animals: Evidence for Hsp104 and Hsp78 chaperones sharing mitochondrial enzymes as clients. PLoS ONE 2015, 10, 1–24, doi:10.1371/journal.pone.0117192.
- Rampelt, H.; Kirstein-Miles, J.; Nillegoda, N.B.; Chi, K.; Scholz, S.R.; Morimoto, R.I.; Bukau, B. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J. 2012, 31, 4221–4235, doi:10.1038/emboj.2012.264.
- Gao, X.; Carroni, M.; Nussbaum-Krammer, C.; Mogk, A.; Nillegoda, N.B.; Szlachcic, A.; Guilbride, D.L.; Saibil, H.R.; Mayer, M.P.; Bukau, B. Human Hsp70 Disaggregase Reverses Parkinson’s-Linked α-Synuclein Amyloid Fibrils. Cell 2015, 59, 781–793, doi:10.1016/j.molcel.2015.07.012.
- Shorter, J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS ONE 2011, 6, e26319, doi:10.1371/journal.pone.0026319.
- Wentink, A.S.; Nillegoda, N.B.; Feufel, J.; Ubartaitė, G.; Schneider, C.P.; de los Rios, P.; Hennig, J.; Barducci, A.; Bukau, B. Molecular dissection of amyloid disaggregation by human HSP70. Nature 2020, 587, 483–488, doi:10.1038/s41586-020-2904-6.
- Roberts, B.T.; Wickner, R.B. Heritable activity: A prion that propagates by covalent autoactivation. Genes Dev. 2003, 17, 2083–2087, doi:10.1101/gad.1115803.
- Zakharov, I.A.; Yarovoy, B.P. Cytoduction as a new tool in studying the cytoplasmic heredity in yeast. Cell. Biochem. 1977, 14, 15–18, doi:10.1007/BF01734159.
- Baxa, U.; Keller, P.W.; Cheng, N.; Wall, J.S.; Steven, A.C. In Sup35p filaments (the [PSI+] prion), the globular C-terminal domains are widely offset from the amyloid fibril backbone. Microbiol. 2011, 79, 523–532, doi:10.1111/j.1365-2958.2010.07466.x.
- Urakov, V.N.; Vishnevskaya, A.B.; Alexandrov, I.M.; Kushnirov, V.V.; Smirnov, V.N.; Ter-Avanesyan, M.D. Interdependence of amyloid formation in yeast: Implications for polyglutamine disorders and biological functions. Prion 2010, 4, 45–52, doi:10.4161/pri.4.1.11074.
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free Formation of RNA Granules: Low Complexity Sequence Domains Form Dynamic Fibers within Hydrogels. Cell 2012, 149, 753–767, doi:10.1016/j.cell.2012.04.017.
- Murakami, T.; Qamar, S.; Lin, J.Q.; Schierle, G.S.K.; Rees, E.; Miyashita, A.; Costa, A.R.; Dodd, R.B.; Chan, F.T.S.; Michel, C.H.; et al. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron 2015, 88, 678–690, doi:10.1016/j.neuron.2015.10.030.
- Yang, Y.-S.; Kato, M.; Wu, X.; Litsios, A.; Sutter, B.M.; Wang, Y.; Hsu, C.-H.; Wood, N.E.; Lemoff, A.; Mirzaei, H.; et al. Yeast Ataxin-2 Forms an Intracellular Condensate Required for the Inhibition of TORC1 Signaling during Respiratory Growth. Cell 2019, 177, 697–710.e17, doi:10.1016/j.cell.2019.02.043.
- Lagaudrière-Gesbert, C.; Newmyer, S.L.; Gregers, T.F.; Bakke, O.; Ploegh, H.L. Uncoating ATPase Hsc70 is recruited by invariant chain and controls the size of endocytic compartments. Natl. Acad. Sci. USA 2002, 99, 1515–1520, doi:10.1073/pnas.042688099.
- Chakravarty, A.K.; Smejkal, T.; Itakura, A.K.; Garcia, D.M.; Jarosz, D.F. A Non-amyloid Prion Particle that Activates a Heritable Gene Expression Program. Cell 2020, 77, 251–265.e9, doi:10.1016/j.molcel.2019.10.028.
- Cox, B.; Ness, F.; Tuite, M. Analysis of the generation and segregation of propagons: Entities that propagate the [PSI+] prion in yeast. Genetics 2003, 165, 23–33.
- Ghaemmaghami, S.; Huh, W.-K.; Bower, K.; Howson, R.W.; Belle, A.; Dephoure, N.; O’Shea, E.K.; Weissman, J.S. Global analysis of protein expression in yeast. Nature 2003, 425, 737–741, doi:10.1038/nature02046.
- Ness, F.; Cox, B.S.; Wongwigkarn, J.; Naeimi, W.R.; Tuite, M.F. Over-expression of the molecular chaperone Hsp104 in Saccharomyces cerevisiae results in the malpartition of [PSI+] propagons. Microbiol. 2017, 104, 125–143, doi:10.1111/mmi.13617.
- Kushnirov, V.V.; Kochneva-Pervukhova, N.V.; Chechenova, M.B.; Frolova, N.S.; Ter-Avanesyan, M.D. Prion properties of the Sup35 protein of yeast Pichia methanolica. EMBO J. 2000, 19, 324–331, doi:10.1093/emboj/19.3.324.
- Gorkovskiy, A.; Reidy, M.; Masison, D.C.; Wickner, R.B. Hsp104 disaggregase at normal levels cures many [PSI +] prion variants in a process promoted by Sti1p, Hsp90, and Sis1p. Natl. Acad. Sci. USA 2017, 114, E4193–E4202, doi:10.1073/pnas.1704016114.
- Moriyama, H.; Edskes, H.K.; Wickner, R.B. [URE3] prion propagation in Saccharomyces cerevisiae: Requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p. Cell. Biol. 2000, 20, 8916–8922.
- Derkatch, I.L.; Bradley, M.E.; Masse, S.V.; Zadorsky, S.P.; Polozkov, G.V.; Inge-Vechtomov, S.G.; Liebman, S.W. Dependence and independence of [PSI+] and [PIN+]: A two-prion system in yeast? EMBO J. 2000, 19, 1942–1952, doi:10.1093/emboj/19.9.1942.
- Matveenko, A.G.; Barbitoff, Y.A.; Jay-Garcia, L.M.; Chernoff, Y.O.; Zhouravleva, G.A. Differential effects of chaperones on yeast prions: CURrent view. Genet. 2018, 64, 317–325, doi:10.1007/s00294-017-0750-3.
- Kushnirov, V.V.; Kryndushkin, D.S.; Boguta, M.; Smirnov, V.N.; Ter-Avanesyan, M.D. Chaperones that cure yeast artificial [PSI+] and their prion-specific effects. Biol. 2000, 10, 1443–1446.
- Greene, L.E.; Saba, F.; Silberman, R.E.; Zhao, X. Mechanisms for curing yeast prions. J. Mol. Sci. 2020, 21, 6536, doi:10.3390/ijms21186536.
- Villali, J.; Dark, J.; Brechtel, T.M.; Pei, F.; Sindi, S.S.; Serio, T.R. Nucleation seed size determines amyloid clearance and establishes a barrier to prion appearance in yeast. Struct. Mol. Biol. 2020, 27, 540–549, doi:10.1038/s41594-020-0416-6.
- Frederick, K.K.; Debelouchina, G.T.; Kayatekin, C.; Dorminy, T.; Jacavone, A.C.; Griffin, R.G.; Lindquist, S. Distinct prion strains are defined by amyloid core structure and chaperone binding site dynamics. Biol. 2014, 21, 295–305, doi:10.1016/j.chembiol.2013.12.013.
- Newnam, G.P.; Birchmore, J.L.; Chernoff, Y.O. Destabilization and recovery of a yeast prion after mild heat shock. Mol. Biol. 2011, 408, 432–448, doi:10.1016/j.jmb.2011.02.034.
- Howie, R.L.; Jay-Garcia, L.M.; Kiktev, D.A.; Faber, Q.L.; Murphy, M.; Rees, K.A.; Sachwani, N.; Chernoff, Y.O. Role of the Cell Asymmetry Apparatus and Ribosome-Associated Chaperones in the Destabilization of a Saccharomyces cerevisiae Prion by Heat Shock. Genetics 2019, 212, 757–771, doi:10.1534/genetics.119.302237.
- Alexandrov, I.M.; Vishnevskaya, A.B.; Ter-Avanesyan, M.D.; Kushnirov, V.V. Appearance and propagation of polyglutamine-based amyloids in yeast: Tyrosine residues enable polymer fragmentation. Biol. Chem. 2008, 283, 15185–15192, doi:10.1074/jbc.M802071200.
- Franco, A.; Gracia, P.; Colom, A.; Camino, J.D.; Fernández-Higuero, J.Á.; Orozco, N.; Dulebo, A.; Saiz, L.; Cremades, N.; Vilar, J.M.G.; et al. All-or-none amyloid disassembly via chaperone-triggered fibril unzipping favors clearance of α-synuclein toxic species. Natl. Acad. Sci. USA 2021, 118, e2105548118, doi:10.1073/pnas.2105548118.
- Shorter, J. Hsp104: A weapon to combat diverse neurodegenerative disorders. NeuroSignals 2007, 16, 63–74, doi:10.1159/000109760.
- Mosser, D.D.; Ho, S.; Glover, J.R. Saccharomyces cerevisiae Hsp104 enhances the chaperone capacity of human cells and inhibits heat stress-induced proapoptotic signaling. Biochemistry 2004, 43, 8107–8115, doi:10.1021/bi0493766.
- Lo Bianco, C.; Shorter, J.; Régulier, E.; Lashuel, H.; Iwatsubo, T.; Lindquist, S.; Aebischer, P. Hsp104 antagonizes alpha-synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. Clin. Invest. 2008, 118, 3087–3097, doi:10.1172/JCI35781.
- Liu, Y.-H.; Han, Y.-L.; Song, J.; Wang, Y.; Jing, Y.-Y.; Shi, Q.; Tian, C.; Wang, Z.-Y.; Li, C.-P.; Han, J.; et al. Heat shock protein 104 inhibited the fibrillization of prion peptide 106-126 and disassembled prion peptide 106-126 fibrils in vitro. J. Biochem. Cell Biol. 2011, 43, 768–774, doi:10.1016/j.biocel.2011.01.022.
- Cushman-Nick, M.; Bonini, N.M.; Shorter, J. Hsp104 suppresses polyglutamine-induced degeneration post onset in a drosophila MJD/SCA3 model. PLoS Genet. 2013, 9, e1003781, doi:10.1371/journal.pgen.1003781.
- Vacher, C.; Garcia-Oroz, L.; Rubinsztein, D.C. Overexpression of yeast hsp104 reduces polyglutamine aggregation and prolongs survival of a transgenic mouse model of Huntington’s disease. Mol. Genet. 2005, 14, 3425–3433, doi:10.1093/hmg/ddi372.
- Satyal, S.H.; Schmidt, E.; Kitagawa, K.; Sondheimer, N.; Lindquist, S.; Kramer, J.M.; Morimoto, R.I. Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Natl. Acad. Sci. USA 2000, 97, 5750–5755, doi:10.1073/pnas.100107297.
- Perrin, V.; Régulier, E.; Abbas-Terki, T.; Hassig, R.; Brouillet, E.; Aebischer, P.; Luthi-Carter, R.; Déglon, N. Neuroprotection by Hsp104 and Hsp27 in lentiviral-based rat models of Huntington’s disease. Ther. 2007, 15, 903–911, doi:10.1038/mt.sj.6300141.
- Mack, K.L.; Shorter, J. Engineering and Evolution of Molecular Chaperones and Protein Disaggregases with Enhanced Activity. Mol. Biosci. 2016, 3, 8, doi:10.3389/fmolb.2016.00008.
- Jackrel, M.E.; Yee, K.; Tariq, A.; Chen, A.I.; Shorter, J. Disparate Mutations Confer Therapeutic Gain of Hsp104 Function. ACS Chem. Biol. 2015, 10, 2672–2679, doi:10.1021/acschembio.5b00765.
- Jackrel, M.E.; Desantis, M.E.; Martinez, B.A.; Castellano, L.M.; Stewart, R.M.; Caldwell, K.A.; Caldwell, G.A.; Shorter, J. Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell 2014, 156, 170–182, doi:10.1016/j.cell.2013.11.047.
- Sweeny, E.A.; Jackrel, M.E.; Go, M.S.; Sochor, M.A.; Razzo, B.M.; DeSantis, M.E.; Gupta, K.; Shorter, J. The Hsp104 N-terminal domain enables disaggregase plasticity and potentiation. Cell 2015, 57, 836–849, doi:10.1016/j.molcel.2014.12.021.
- Tariq, A.; Lin, J.; Noll, M.M.; Torrente, M.P.; Mack, K.L.; Murillo, O.H.; Jackrel, M.E.; Shorter, J. Potentiating Hsp104 activity via phosphomimetic mutations in the middle domain. FEMS Yeast Res. 2018, 18, foy042, doi:10.1093/femsyr/foy042.
- Tariq, A.; Lin, J.; Jackrel, M.E.; Hesketh, C.D.; Carman, P.J.; Mack, K.L.; Weitzman, R.; Gambogi, C.; Hernandez Murillo, O.A.; Sweeny, E.A.; et al. Mining Disaggregase Sequence Space to Safely Counter TDP-43, FUS, and α-Synuclein Proteotoxicity. Cell Rep. 2019, 28, 2080–2095.e6, doi:10.1016/j.celrep.2019.07.069.
- Ryan, J.J.; Bao, A.; Bell, B.; Ling, C.; Jackrel, M.E. Drivers of Hsp104 potentiation revealed by scanning mutagenesis of the middle domain. Protein Sci. 2021, 30, 1667–1685, doi:10.1002/pro.4126.
- Reidy, M.; Miot, M.; Masison, D.C. Prokaryotic Chaperones Support Yeast Prions and Thermoto lerance and Define Disaggregation Machinery Interactions. Genetics 2012, 192, 185–193, doi:10.1534/genetics.112.142307.
- March, Z.M.; Sweeney, K.; Kim, H.; Yan, X.; Castellano, L.M.; Jackrel, M.E.; Lin, J.; Chuang, E.; Gomes, E.; Willicott, C.W.; et al. Therapeutic genetic variation revealed in diverse Hsp104 homologs. Elife 2020, 9, e57457, doi:10.7554/eLife.57457.
- Michalska, K.; Zhang, K.; March, Z.M.; Hatzos-Skintges, C.; Pintilie, G.; Bigelow, L.; Castellano, L.M.; Miles, L.J.; Jackrel, M.E.; Chuang, E.; et al. Structure of Calcarisporiella thermophila Hsp104 Disaggregase that Antagonizes Diverse Proteotoxic Misfolding Events. Structure 2019, 27, 449–463.e7, doi:10.1016/j.str.2018.11.001.
- Cupo, R.R.; Shorter, J. Skd3 (human ClpB) is a potent mitochondrial protein disaggregase that is inactivated by 3-methylglutaconic aciduria-linked mutations. Elife 2020, 9, e55279, doi:10.7554/eLife.55279.
- Smith, D.M.; Benaroudj, N.; Goldberg, A. Proteasomes and their associated ATPases: A destructive combination. Struct. Biol. 2006, 156, 72–83, doi:10.1016/j.jsb.2006.04.012.
- Brooks, C.; Snoberger, A.; Belcastro, M.; Murphy, J.; Kisselev, O.G.; Smith, D.M.; Sokolov, M. Archaeal Unfoldase Counteracts Protein Misfolding Retinopathy in Mice. Neurosci. 2018, 38, 7248–7254, doi:10.1523/JNEUROSCI.0905-18.2018.
- Tittelmeier, J.; Sandhof, C.A.; Ries, H.M.; Druffel-Augustin, S.; Mogk, A.; Bukau, B.; Nussbaum-Krammer, C. The HSP110/HSP70 disaggregation system generates spreading-competent toxic α-synuclein species. EMBO J. 2020, 39, e103954, doi:10.15252/embj.2019103954.
- Demaegd, K.; Schymkowitz, J.; Rousseau, F. Transcellular Spreading of Tau in Tauopathies. Chembiochem 2018, 19, 2424–2432, doi:10.1002/cbic.201800288.
This entry is adapted from the peer-reviewed paper 10.3390/biom11121884