The chronic infection established by the human immunodeficiency virus 1 (HIV-1) produces serious CD4+ T cell immunodeficiency despite the decrease in HIV-1 ribonucleic acid (RNA) levels and the raised life expectancy of people living with HIV-1 (PLWH) through treatment with combined antiretroviral therapies (cART). HIV-1 enters the central nervous system (CNS), where perivascular macrophages and microglia are infected. Serious neurodegenerative symptoms related to HIV-associated neurocognitive disorders (HAND) are produced by infection of the CNS. Despite advances in the treatment of this infection, HAND significantly contribute to morbidity and mortality globally.
- virus
- HAND
- central nervous system
1. Introduction
HIV-associated neurological disease can produce significant morbidity and mortality globally and can be due to HIV replication, opportunistic infections, or comorbidities [1] that can structurally and functionally affect the brain [2]; this affection can occur in a primary or secondary way, also affecting meninges and muscles, among others [3]. HIV-1 infection can produce neurocognitive damage and motor disorders [4]. This infection can be considered as a mix of virus-related neurocognitive diseases and neuronal tissue inflammation [4][5]. The use of cART with an improved ability to penetrate the BBB has drastically diminished the occurrence of these problems [5][6]. Nevertheless, since not all anti-HIV drugs are capable of crossing the BBB with effectiveness, viral reservoirs still persist, causing neurological disorders [7]. As a result, these clinical symptoms still persist as crucial problems for people living with HIV (PLWH), particularly for children or patients with low adherence to treatment [8], the latter becoming increasingly frequent among old PLWH since age is a risk factor for functional impairment and disability [9]. Other crucial risk factors are low levels of CD4+ T cells, an increase in plasma viral load, hepatitis C virus (HCV) coinfection, and metabolic comorbidities in PLWH with neurocognitive impairment [4][6]. Taking into account the restrictions of current therapeutic strategies, novel approaches are a key point in the neuro-HIV research. Some of these methods involve gene therapy, gene editing, RNA interference, and modulation of different cellular physiological processes (reviewed in Ojha et al., 2017, and Kwarteng et al., 2017) [10][11]. Even though the precise etiology of HIV-associated neurocognitive disorders (HAND) in the cART era has not yet been deeply studied, permanent inflammation in the CNS is a frequent characteristic of this disease that can lead to neurocognitive impairment [12].
2. HAND
2.1. Advances and Evolution of the Antiretroviral Treatment of HIV Infection
| Antiretroviral Drug Class | 4 (Very Good) | 3 (Good) | 2 (Fair) | 1 (Poor) |
|---|---|---|---|---|
| Nucleoside Reverse Transcriptase Inhibitors (NRTIs) | Zidovudine | Abacavir Emtricitabine |
Didanosine Lamivudine Stavudine |
Adefovir Tenofovir Zalcitabine |
| Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs) | Nevirapine | Delavirdine Efavirenz |
Etravirine | |
| Protease Inhibitors (PIs) | Amprenavir-r Indinavir-r |
Amprenavir Danmavir Darunavir Fosamprenavir-r Indinavir Lopinavir-r |
Atazanavir Atazanarir-r Fosamprenavir |
Nelfinavir Ritonavir Saquinavir Saquinavir-r Tipranavir-r |
| Integrase Inhibitors | Elvitegravir Raltegravir |
|||
| Entry Inhibitors | Maraviroc Vicriviroc |
Enfuvirfide T-1249 |
2.2. Classification of HAND
2.3. Pathology of NeuroAIDS
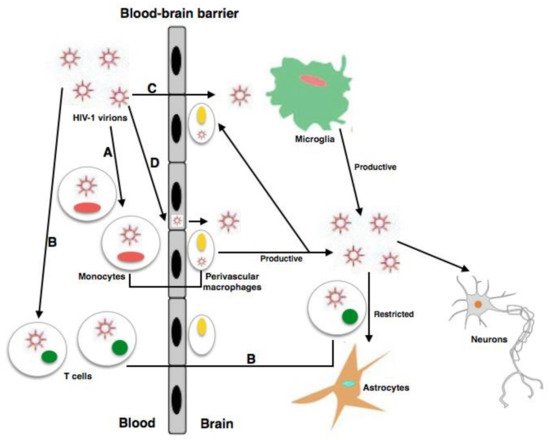
2.4. HIV Viral Proteins Involved in Neuropathogenesis
2.5. Inflammation and Role of Mononuclear Phagocytes in HAND
This entry is adapted from the peer-reviewed paper 10.3390/microorganisms9122537
References
- Chai, Q.; Jovasevic, V.; Malikov, V.; Sabo, Y.; Morham, S.; Walsh, D.; Naghavi, M.H. Yes, it is time to reconsider how we rate cognitive impairments in HIV disease. Neuroepidemiology 2013, 41, 217–218.
- Thakur, K.T.; Boubour, A.; Saylor, D.; Das, M.; Bearden, D.R.; Birbeck, G.L. Global HIV neurology: A comprehensive review. AIDS 2019, 33, 163–184.
- Rosenthal, J.; Tyor, W. Aging, comorbidities, and the importance of finding biomarkers for HIV-associated neurocognitive disorders. J. Neurovirol. 2019, 5, 673–685.
- Scutari, R.; Alteri, C.; Perno, C.F.; Svicher, V.; Aquaro, S. The Role of HIV Infection in Neurologic Injury. Brain Sci. 2017, 7, 38.
- Aquaro, S.; Panti, S.; Caroleo, M.C.; Balestra, E.; Cenci, A.; Forbici, F. Primary macrophages infected by human immunodeficiency virus trigger CD95-mediated apoptosis of uninfected astrocytes. J. Leukoc. Biol. 2000, 68, 429–435.
- Sacktor, N. Changing clinical phenotypes of HIV-associated neurocognitive disorders. J. Neurovirol. 2018, 24, 141–145.
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder--pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248, Erratum in Nat. Rev. Neurol. 2016, 12, 308.
- DeVaughn, S.; Müller-Oehring, E.M.; Markey, B.; Brontë-Stewart, H.M.; Schulte, T. Aging with HIV-1 Infection: Motor Functions, Cognition, and Attention—A Comparison with Parkinson’s Disease. Neuropsychol. Rev. 2015, 25, 424–438.
- Khalili, K.; White, M.K.; Jacobson, J.M. Novel AIDS therapies based on gene editing. Cell Mol. Life Sci. 2017, 74, 2439–2450.
- Ojha, C.R.; Lapierre, J.; Rodriguez, M.; Dever, S.M.; Zadeh, M.A.; DeMarino, C.; Pleet, M.L.; Kashanchi, F.; El-Hage, N. Interplay between Autophagy, Exosomes and HIV-1 Associated Neurological Disorders: New Insights for Diagnosis and Therapeutic Applications. Viruses 2017, 9, 176.
- Kwarteng, A.; Ahuno, S.T.; Kwakye-Nuako, G. The therapeutic landscape of HIV-1 via genome editing. AIDS Res. Ther. 2017, 14, 32.
- Marban, C.; Forouzanfar, F.; Ait-Ammar, A.; Fahmi, F.; El Mekdad, H.; Daouad, F.; Rohr, O.; Schwartz, C. Targeting the Brain Reservoirs: Toward an HIV Cure. Front. Immunol. 2016, 7, 397.
- Svicher, V.; Marchetti, G.; Ammassari, A.; Ceccherini-Silberstein, F.; Sarmati, L. Impact Study Group. Novelties in Evaluation and Monitoring of Human Immunodeficiency Virus-1 Infection: Is Standard Virological Suppression Enough for Measuring Antiretroviral Treatment Success? AIDS Rev. 2017, 19, 119–133.
- Ding, Y.; Duan, S.; Ye, R.; Yang, Y.; Yao, S.; Wang, J.; Cao, D.; Liu, X.; Lu, L.; Jia, M. More improvement than progression of liver fibrosis following antiretroviral therapy in a longitudinal cohort of HIV-infected patients with or without HBV and HCV co-infections. J. Viral Hepat. 2017, 24, 412–420.
- Tsegaw, M.; Andargie, G.; Alem, G.; Tareke, M. Screening HIV-associated neurocognitive disorders (HAND) among HIV positive patients attending antiretroviral therapy in South Wollo, Ethiopia. J. Psychiatr Res. 2017, 85, 37–41.
- Cole, J.H.; Underwood, J.; Caan, M.W.; De Francesco, D.; Van Zoest, R.A.; Leech, R.; Wit, F.W.; Portegies, P.; Geurtsen, G.; Schmand, B.A.; et al. Increased brain-predicted aging in treated HIV disease. Neurology 2017, 88, 1349–1357.
- Hellmuth, J.; Milanini, B.; Valcour, V. Interactions between ageing and NeuroAIDS. Curr. Opin. HIV AIDS 2014, 9, 527–532.
- Brew, B.J.; Crowe, S.M.; Landay, A.; Cysique, L.; Guillemin, G. Neurodegeneration and ageing in the HAART era. J. Neuroimmune Pharmacol. 2009, 4, 163–174.
- Sacktor, N.; Skolasky, R.L.; Moxley, R.; Wang, S.; Mielke, M.M.; Munro, C.; Steiner, J.; Nath, A.; Haughey, N.; McArthur, J. Paroxetine and fluconazole therapy for HIV-associated neurocognitive impairment: Results from a double-blind, placebo-controlled trial. J. Neurovirol. 2017, 24, 16–27.
- Gates, T.M.; Cysique, L.; Siefried, K.J.; Chaganti, J.; Moffat, K.J.; Brew, B. Maraviroc-intensified combined antiretroviral therapy improves cognition in virally suppressed HIV-associated neurocognitive disorder. AIDS 2016, 30, 591–600.
- Saloner, R.; Cysique, L.A. HIV-Associated Neurocognitive Disorders: A Global Perspective. J. Int. Neuropsychol. Soc. 2017, 23, 860–869.
- Aquaro, S.; Borrajo, A.; Pellegrino, M.; Svicher, V. Mechanisms underlying of antiretroviral drugs in different cellular reservoirs with a focus on macrophages. Virulence 2020, 11, 400–413.
- Kim, B.-H.; Kelschenbach, J.; Borjabad, A.; Hadas, E.; He, H.; Potash, M.J.; Nedelcovych, M.T.; Rais, R.; Haughey, N.J.; McArthur, J.C.; et al. Intranasal insulin therapy reverses hippocampal dendritic injury and cognitive impairment in a model of HIV-associated neurocognitive disorders in EcoHIV-infected mice. AIDS 2019, 33, 973–984.
- Borrajo, A.; Spuch, C.; Penedo, M.A.; Olivares, J.M.; Agís-Balboa, R.C. Important role of microglia in HIV-1 associated neurocognitive disorders and the molecular pathways implicated in its pathogenesis. Ann. Med. 2021, 53, 43–69.
- Bandera, A.; Taramasso, L.; Bozzi, G.; Muscatello, A.; Robinson, J.A.; Burdo, T.H.; Gori, A. HIV-Associated Neurocognitive Impairment in the Modern ART Era: Are We Close to Discovering Reliable Biomarkers in the Setting of Virological Suppression? Front. Aging Neurosci. 2019, 11, 187.
- Eggers, C.; Arendt, G.; Hahn, K.; Husstedt, I.W.; Maschke, M.; Neuen-Jacob, E.; Obermann, M.; Rosenkranz, T.; Schielke, E. HIV-1-associated neurocognitive disorder: Epidemiology, pathogenesis, diagnosis, and treatment. J. Neurol. 2017, 264, 1715–1727.
- Liu, N.Q.; Lossinsky, A.S.; Popik, W.; Li, X.; Gujuluva, C.; Kriederman, B.; Roberts, J.; Pushkarsky, T.; Bukrinsky, M.; Witte, M.; et al. Human Immunodeficiency Virus Type 1 Enters Brain Microvascular Endothelia by Macropinocytosis Dependent on Lipid Rafts and the Mitogen-Activated Protein Kinase Signaling Pathway. J. Virol. 2002, 76, 6689–6700.
- Cho, Y.E.; Lee, M.H.; Song, B.J. Neuronal cell death and degeneration through increased nitroxidative stress and tau phosphorylation in HIV-1 transgenic rats. PLoS ONE 2017, 12, e0169945.
- Torres-Munoz, J.; Stockton, P.; Tacoronte, N.; Roberts, B.; Maronpot, R.R.; Petito, C.K. Detection of HIV-1 gene sequences in hippocampal neurons isolated from postmortem AIDS brains by laser capture microdissection. J. Neuropathol. Exp. Neurol. 2001, 60, 885–892.
- Torres-Munoz, J.E.; Nunez, M.; Petito, C.K. Successful application of hyperbranched multidisplacement genomic amplification to detect HIV-1 sequences in single neurons removed from autopsy brain sections by laser capture microdissection. J. Mol. Diagn. 2008, 10, 317–324.
- Sturdevant, C.B.; Joseph, S.B.; Schnell, G.; Price, R.W.; Swanstrom, R.; Spudich, S. Compartmentalized Replication of R5 T Cell-Tropic HIV-1 in the Central Nervous System Early in the Course of Infection. PLoS Pathog. 2015, 11, e1004720.
- Mukhtar, M.; Acheampong, E.; Khan, M.A.; Bouhamdan, M.; Pomerantz, R.J. Down-modulation of the CXCR4 co-receptor by intracellular expression of a single chain variable fragment (Sf) inhibits HIV-1 entry into primary human brain microvascular endothelial cells and post-mitotic neurons. Brain Res. Mol. Brain Res. 2005, 135, 48–57.
- Cantó-Nogués, C.; Sánchez-Ramón, S.; Álvarez, S.; Lacruz, C.; Muñóz-Fernández, M.Á. HIV-1 infection of neurons might account for progressive HIV-1-associated encephalopathy in children. J. Mol. Neurosci. 2005, 27, 79–89.
- Wang, X.; Zhou, Y.; Gao, Q.; Ping, D.; Wang, Y.; Wu, W.; Lin, X.; Fang, Y.; Zhang, J.; Shao, A. The Role of Exosomal microRNAs and Oxidative Stress in Neurodegenerative Diseases. Oxidative Med. Cell. Longev. 2020, 1–17.
- Rao, V.R.; Ruiz, A.P.; Prasad, V.R. Viral and cellular factors underlying neuropathogenesis in HIV associated neurocognitive disorders (HAND). AIDS Res. Ther. 2014, 11, 13.
- Haughey, N.J.; Nath, A.; Mattson, M.P.; Slevin, J.T.; Geiger, J.D. HIV-1 tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J. Neurochem. 2001, 78, 457–467.
- Zhou, L.; Saksena, N.K. HIV associated neurocognitive disorders. Infect. Dis. Rep. 2013, 5, e8.
- Rozzi, S.J.; Avdoshina, V.; Fields, J.A.; Mocchetti, I. Human immunodeficiency virus Tat impairs mitochondrial fission in neurons. Cell Death Discov. 2018, 4, 1–12.
- Kanmogne, G.D.; Primeaux, C.; Grammas, P. HIV-1 gp120 proteins alter tight junction protein expression and brain endothelial cell permeability: Implications for the pathogenesis of HIV-associated dementia. J. Neuropathol. Exp. Neurol. 2005, 64, 498–505.
- Avdoshina, V.; Taraballi, F.; Dedoni, S.; Corbo, C.; Paige, M.; Saygideğer Kont, Y.; Üren, A.; Tasciotti, E.; Mocchetti, I. Identification of a binding site of the human immunodeficiency virus envelope protein gp120 to neuronal-specific tubulin. J. Neurochem. 2016, 137, 287–298.
- Wayman, W.N.; Dodiya, H.B.; Persons, A.L.; Kashanchi, F.; Kordower, J.H.; Hu, X.-T.; Napier, T.C. Enduring cortical alterations after a single in vivo treatment of HIV-1 Tat. NeuroReport 2012, 23, 825–829.
- Berman, J.W.; Carvallo, L.; Buckner, C.M.; Luers, A.; Prevedel, L.; Bennett, M.V.; Eugenin, E.A. HIV-tat alters Connexin43 expression and trafficking in human astrocytes: Role in NeuroAIDS. J. Neuroinflammation 2016, 13, 54.
- Shin, A.H.; Thayer, S.A. Human immunodeficiency virus-1 protein Tat induces excitotoxic loss of presynaptic terminals in hippocampal cultures. Mol. Cell. Neurosci. 2013, 54, 22–29.
- Zucchini, S.; Pittaluga, A.; Brocca-Cofano, E.; Summa, M.; Fabris, M.; De Michele, R.; Bonaccorsi, A.; Busatto, G.; Barbanti-Brodano, G.; Altavilla, G.; et al. Increased excitability in tat-transgenic mice: Role of tat in HIV-related neurological disorders. Neurobiol. Dis. 2013, 55, 110–119.
- Midde, N.M.; Gomez, A.; Zhu, J. HIV-1 Tat Protein Decreases Dopamine Transporter Cell Surface Expression and Vesicular Monoamine Transporter-2 Function in Rat Striatal Synaptosomes. J. Neuroimmune Pharmacol. 2012, 7, 629–639.
- Laforge, M.; Petit, F.; Estaquier, J.; Senik, A. Commitment to Apoptosis in CD4 + T Lymphocytes Productively Infected with Human Immunodeficiency Virus Type 1 Is Initiated by Lysosomal Membrane Permeabilization, Itself Induced by the Isolated Expression of the Viral Protein Nef. J. Virol. 2007, 81, 11426–11440.
- Accession, M.Y.; Hasselrot, U.; Wu, G.; Nath, A.; Anderson, C.; Mactutus, C.F. Temporal relationships between HIV-1 Tat-induced neuronal degeneration, OX-42 immunoreactivity, reactive astrocytosis, and protein oxidation in the rat striatum. Brain Res. 2003, 987, 1–9.
- Acheampong, E.A.; Parveen, Z.; Muthoga, L.W.; Wasmuth-Peroud, V.; Kalayeh, M.; Bashir, A.; Diecidue, R.; Mukhtar, M.; Pomerantz, R.J. Molecular Interactions of Human Immunodeficiency Virus Type 1 with Primary Human Oral Keratinocytes. J. Virol. 2005, 79, 8440–8453.
- Gavriil, E.S.; Cooney, R.; Weeks, B.S. Tat Mediates Apoptosis in Vivo in the Rat Central Nervous System. Biochem. Biophys. Res. Commun. 2000, 267, 252–256.
- Jimenez-Guardeño, J.M.; Apolonia, L.; Betancor, G.; Malim, M.H. Immunoproteasome activation enables human TRIM5α restriction of HIV-1. Nat. Microbiol. 2019, 4, 933–940.
- André, P.; Groettrup, M.; Klenerman, P.; de Giuli, R.; Booth, B.L.; Cerundolo, V.; Bonneville, M.; Jotereau, F.; Zinkernagel, R.M.; Lotteau, V. An inhibitor of HIV-1 protease modulates proteasome activity, antigen presentation, and T cell responses. Proc. Natl. Acad. Sci. USA 1998, 95, 13120–13124.
- Ancuta, P.; Kamat, A.; Kunstman, K.J.; Kim, E.-Y.; Autissier, P.; Wurcel, A.; Zaman, T.; Stone, D.; Mefford, M.; Morgello, S.; et al. Microbial Translocation Is Associated with Increased Monocyte Activation and Dementia in AIDS Patients. PLoS ONE 2008, 3, e2516.
- Gray, L.R.; Cowley, D.; Welsh, C.; Lu, H.K.; Brew, B.; Lewin, S.R.; Wesselingh, S.L.; Gorry, P.R.; Churchill, M.J. CNS-specific regulatory elements in brain-derived HIV-1 strains affect responses to latency-reversing agents with implications for cure strategies. Mol. Psychiatry 2015, 21, 574–584.
- Zevin, A.S.; McKinnon, L.; Burgener, A.; Klatt, N.R. Microbial translocation and microbiome dysbiosis in HIV-associated immune activation. Curr. Opin. HIV AIDS 2016, 11, 182–190.
- Lutgen, V.; Narasipura, S.D.; Barbian, H.J.; Richards, M.; Wallace, J.; Razmpour, R.; Buzhdygan, T.; Ramirez, S.; Prevedel, L.; Eugenin, E.A.; et al. HIV infects astrocytes in vivo and egresses from the brain to the periphery. PLoS Pathog. 2020, 16, e1008381.
- Brown, D.G.; Soto, R.; Yandamuri, S.; Stone, C.; Dickey, L.; Gomes-Neto, J.C.; Pastuzyn, E.D.; Bell, R.; Petersen, C.; Buhrke, K.; et al. The microbiota protects from viral-induced neurologic damage through microglia-intrinsic TLR signaling. eLife 2019, 8, e47117.
- Spudich, S.S. Immune activation in the central nervous system throughout the course of HIV infection. Curr. Opin. HIV AIDS 2016, 11, 226–233.
- Prinz, M.; Tay, T.L.; Wolf, Y.; Jung, S. Microglia: Unique and common features with other tissue macrophages. Acta Neuropathol. 2014, 128, 319–331.
- Logsdon, A.F.; Erickson, M.; Rhea, E.M.; Salameh, T.S.; Banks, W.A. Gut reactions: How the blood–brain barrier connects the microbiome and the brain. Exp. Biol. Med. 2017, 243, 159–165.
- Clarke, T.B.; Davis, K.M.; Lysenko, E.S.; Zhou, A.Y.; Yu, Y.; Weiser, J.N. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat. Med. 2010, 16, 228–231.
- Bohórquez, D.V.; Shahid, R.A.; Erdmann, A.; Kreger, A.M.; Wang, Y.; Calakos, N.; Wang, F.; Liddle, R.A. Neuroepithelial circuit formed by innervation of sensory enteroendocrine cells. J. Clin. Investig. 2015, 125, 782–786.
- Shaw, T.N.; Houston, S.A.; Wemyss, K.; Bridgeman, H.M.; Barbera, T.A.; Zangerle-Murray, T.; Strangward, P.; Ridley, A.; Wang, P.; Tamoutounour, S.; et al. Tissue-resident macrophages in the intestine are long lived and defined by Tim-4 and CD4 expression. J. Exp. Med. 2018, 215, 1507–1518.
- Wong, S.W.; Kwon, M.-J.; Choi, A.M.; Kim, H.-P.; Nakahira, K.; Hwang, D.H. Fatty Acids Modulate Toll-like Receptor 4 Activation through Regulation of Receptor Dimerization and Recruitment into Lipid Rafts in a Reactive Oxygen Species-dependent Manner. J. Biol. Chem. 2009, 284, 27384–27392.