Clonal hematopoiesis (CH), a process that involves the accumulation of somatic mutations in hematopoietic stem cells which leads to clonal expansion of mutations in blood cells, may account for the non-tumor derived mutations detected from plasma. These CH mutations may act as a biological noise to cfDNA analysis and complicate the interpretation of mutations detected from liquid biopsy.
- liquid biopsy
- circulating tumor DNA
- clonal hematopoiesis
- next-generation sequencing
1. Background
In recent years, liquid biopsy, which involves genomic profiling of tumors using circulating biomarkers in the bodily fluid, has attracted tremendous interest in the field of cancer diagnosis and management [1]. The accessibility and low invasiveness of blood sampling compared to tumor tissue biopsy received great interest for their potential uses in various clinical applications. Liquid biopsy comprises several circulating tumor circulomes, including, circulating tumor proteins, circulating tumor cells, circulating tumor nucleic acid (DNA and RNA), extracellular vesicles, and tumor-educated platelets [2]. Recent advancement in sequencing technology and bioinformatics allowed accurate detection of genetic alterations in circulating tumor DNA (ctDNA) from the blood. ctDNA is highly degraded DNA fragments released from tumor cells and they recapitulate the tumor’s molecular alterations [3]. However, there are also limitations to this approach. Owing to the complex nature of blood plasma, alterations detected from cell-free DNA (cfDNA) could be tumor-derived or non-tumor derived. Maintaining the sensitivity and specificity to detect true tumor-derived cfDNA from plasma remains the biggest challenge for its routine use in clinical practice. The majority of cfDNA present in the blood are derived from hematopoietic cells [4].
Clonal hematopoiesis (CH), a process that involves the accumulation of somatic mutations in hematopoietic stem cells which leads to clonal expansion of mutations in blood cells, may account for the non-tumor derived mutations detected from plasma. CH is part of the normal process of aging, and they are highly prevalent in the general population [5][6]. These mutations from hematopoietic cells which could disguise as tumor-derived, often present as a source of biological background noise to cfDNA analysis. Incorrect classification of mutations detected in cfDNA analysis as tumor-associated mutations could lead to inappropriate therapeutic decisions for patient management. Furthermore, CH mutations have been reported with several points of clinical significance. Healthy individuals who are carriers of CH have shown an increased risk for developing cardiovascular diseases and hematological malignancies while cancer patients with CH mutations are more likely to develop therapy-related myeloid neoplasms several years after the completion of chemotherapy [5][7]. These observed pathological associations highlight the potential clinical importance of CH detected from liquid biopsy, therefore, CH should not be merely perceived as a source of biological background noise to cfDNA analysis.
2. Clonal Hematopoiesis
2.1. Definition of Clonal Hematopoiesis (CH)
Studies of X-chromosome inactivation in the early 1990s led to the discovery that clonal expansion of blood cells was not only occurring in hematological malignancies but also in healthy individuals as a result of aging [8][9]. It has been estimated that each hematopoietic stem cell acquires one exonic mutation per decade of a normal healthy individual’s life. Based on the estimation that an adult human has an approximately 50,000 to 200,000 stem cells [10], an average person would potentially harbor up to 1.2 million exonic mutations by the age of 70 [6]. The hematopoietic cells with naturally occurring mutations may have the advantage of expanding more rapidly than non-mutated cells; this process is also known as CH [6]. Many reputable studies have been conducted in the past decade to explore CH and different terminologies and definitions were used. It is now generally accepted that the term “CH” refers to any clonal outgrowth of hematopoietic cells, regardless of cause or disease state, while clonal hematopoiesis of indeterminate potential or CHIP usually refers to mutations in driver genes known to be associated with hematological malignancies. CHIP is detected in the DNA of white blood cells from individuals without any symptoms or clinical presentations of malignancy [11]. These mutations will also need to be detected with a minimum variant allele frequency (VAF) of 2% to be classified as CHIP [11]. The terms CH and CHIP will also be used with the definitions mentioned above throughout this review.
2.2. Clinical Implications of CH
2.2.1. Hematological Cancer
The presence of CH does not necessarily indicate hematological malignancies or any alterations in the blood cell counts that reflect the clinical presentation of malignancy. However, the association between CHIP and the risk of developing hematological malignancies has been well documented in previous studies [5][12][13][14][15]. In population-based studies that were followed up for several years, there was a 2–13 fold increase in the relative risk of developing hematological malignancies in individuals that harbored CHIP [5][12][13][14]. Although the relative risk of developing hematological malignancies in individuals with CHIP is significant, the absolute risk remained low. It is estimated that approximately 0.5–1% of CHIP cases would progress to malignancy per year [5][12]. However, individuals having CH mutations with higher frequencies (VAF > 10%) were fivefold more likely to develop hematological malignancies compared to those having mutations with lower VAF [5][15]. Large population case-control studies with 10 years of follow-up improved the understanding of the association between CHIP and AML [14][15]; mutations in genes such as TP53, IDH1, and IDH2 demonstrated increased specificity and penetrance for the development of AML. However, many of the individuals who carried mutations in these genes also harbored mutations in other driver genes, such as NPM1 and FLT3 [5][12][14]. The mutations in these two driver genes are often absent before the development of AML [5][12][14]. These observations suggest that CH itself is likely to be insufficient to induce malignancy and mutations in additional driver genes are required for the development of hematologic malignancy. Further studies are required to evaluate the potential use of CH mutations for risk assessment of hematological malignancies. Since CH mutations originate mostly from hematopoietic stem cells which differentiate to the full spectrum of hematopoietic cells, it is theoretically possible that CH could predispose to any type of hematological malignancies. However, most conducted studies have only observed the association of CH with myeloid malignancies. Future studies with large cohorts should investigate the association of lymphoid neoplasms and CH [16].
2.2.2. Therapy-Related Myeloid Neoplasms (t-MN)
Several studies have suggested that cancer patients who carry CHIP mutations before the initiation of chemotherapy are more likely to develop t-MN compared to those with no CHIP [24]. It is believed that exposure to chemotherapy acts as an external pressure to favor the survival of hematopoietic stem cells with CHIP and has the advantage of outgrowing those cells without CHIP. Notably, an increasing number of CHIP mutations with high VAF are associated with an increased risk of developing t-MN. The mutational spectrum in patients who developed t-MN had a higher prevalence of mutations in DNA damage response genes such as TP53 [25]. The early acquisition of TP53 mutations in CH contributes to the poor responses to chemotherapy seen in patients with t-AML/t-MDS. The clones with CH TP53 mutations are most likely to be resistant to chemotherapy and expand as a result of selective pressure [25]. Future large populational studies are required to expand our current understanding of CH and therapy-related hematological malignancies to accurately predict which individual with CHIP would proceed to develop malignancies.
2.2.3. Cardiovascular Disease (CVD)
The association between CH and human disease is not limited to cancer. Several studies have found that CHIP carriers were 2–4 times more prone to developing coronary heart diseases including myocardial infarction and ischemic stroke than those without CHIP [5][26][27]. In particular, one recent study (the largest so far) conducted exome sequencing of over 35,000 individuals without previous cardiovascular diseases[27]. The study identified individuals with DNMT3A and TET2 CHIP had an increased risk of cardiovascular disease compared to non-carriers after nearly 7 years of follow-up [27]. Furthermore, the authors of this study observed that CHIP carriers with genotypes of reduced IL-6 signaling abrogated the risk of CVD. Similar to hematological malignancies, the greater risk was also observed in individuals who harbor CHIP with VAF greater than 10% [27]. TET2, ASXL1, JAK2, and DNMT3A are the most commonly detected CH mutations in CVD patients [26][28]. Particularly, in individuals bearing JAK2-V617F, the relative risk of coronary heart disease was 12 times higher compared to non-carriers, which was also much higher compared to mutations in other genes [26]. CHIP carriers with pre-existing CVD have also been shown to have worse survival outcomes and increased disease progression than those without CHIP [28]. The profound effect of CH mutations on the prognosis of CVD was also observed in patients with severe calcified aortic valve stenosis who underwent transcatheter aortic valve implantation [29]. The mortality rate in patients with CH mutations was nearly three times higher compared to the non-carriers [29].
3. CH in Liquid Biopsy
3.1. Detection of CH from Plasma
A handful of studies have been conducted in the past few years to assess the detection of CH mutations in plasma and their impact on the interpretation of blood liquid biopsy results (Table 1). The hypothesis of the detection of CH mutations in plasma cfDNA was first suggested in two exploratory studies in small-cell lung cancer (SCLC) and NSCLC patients [22][30]. A total of 5–15% of TP53 mutations detected in the plasma cfDNA of lung cancer patients were also detected in white blood cells, suggesting their CH origin. These early observations were validated in a recent prospective study that performed deep sequencing of cfDNA and matched white blood cells over 124 patients with metastatic cancer using a large gene panel (508 genes) [31]. Close to 50% of the mutations detected in plasma cfDNA were also detected in patient-matched white blood cells. Similar detection rates of CH mutation from plasma cfDNA was also reported in early-stage NSCLC patients [32]. In addition, paired sequencing of plasma cfDNA and white blood cells from healthy individuals showed that the vast majority of mutations (66–90%) detected in plasma cfDNA were originated from CH [24][30][31][32]. The sequencing gene panel, sequencing depth, and the resulting limit of detection varied among these studies, therefore, it is difficult to directly compare the results among the studies. However, the key message is that CH mutations can contribute greatly to the mutations detected from liquid biopsy and clear assessment should be made to identify tumor-derived cfDNA from plasma samples.
Table 1. Summary of published studies on the detection of CH mutations from plasma cell-free DNA (cfDNA) analysis.
|
Cancer Type |
Stage |
Study Size |
Gene Panel |
Depth * |
Reported LOD (%) |
Prevalence of CH Detection |
Study |
|
SCLC |
I–IV |
SCLC: 51 |
TP53 |
NR |
NR |
SCLC: 5.3% |
Fernandez-Cuesta et al., 2016 [30] |
|
Cancer-free |
- |
821 |
50 genes |
40,000x |
0.10% |
89% |
Xia et al., 2017 [33] |
|
NSCLC |
III–IV |
122 |
Focused on TP53 analysis |
NR |
NR |
15% |
Hu et al., 2018 [22] |
|
Prostate |
IV |
217 |
305 genes |
814x |
1% |
15% |
Mayrhofer et al., 2018 [36] |
|
Cancer-free |
- |
259 |
599 genes |
cfDNA: 6200x |
0.25% |
66% |
Liu et al., 2019 [18] |
|
Various solid tumors |
IV |
Cancer:124 |
508 genes |
60,000x |
0.1% |
Cancer: 53% |
Razavi et al., 2019 [31] |
|
Gastric |
I–IV |
788 |
58 genes |
30,000x |
0.1% |
44% |
Leal et al., 2020 [35] |
|
Renal cell carcinoma |
IV |
55 |
981 genes |
Collapsed: 938x |
1% |
20% |
Bacon et al., 2020 [40] |
|
NSCLC |
I–III |
NSCLC: 104 |
255 genes |
Collapsed: 4000–5000x |
0.01% |
NSCLC: 58% |
Chabon et al., 2020 [32] |
|
CRC |
I–IV |
38 |
52 genes |
48,000x |
0.1% |
17% |
Chan et al., 2020 [34] |
* Refers to raw sequencing depth unless otherwise stated; NR: not reported, SCLC- small cell lung cancer; NSCLC- non-small cell lung cancer; CRC- colorectal cancer; LOD- limit of detection.
CH mutations detected from plasma are similar to the mutations detected from white blood cells, involved both canonical CH genes, DNMT3A, TET2, ASXL1 and JAK2, and actionable mutations in genes related to solid tumors, KRAS, PIK3CA and EGFR [18][22][31][33]. Majority of these identified CH variants have been previously reported as tumor-associated somatic mutations which complicate the curation of the variants detected in cfDNA analysis [34][35]. Further stratification to focus on the most frequently mutated TP53 variants in CH showed these hotspot variants coincide with the most frequently mutated TP53 variants in solid tumors (Figure 1). VAF of CH mutations detected from plasma are highly correlated and indifferent to the VAF detected in white blood cells [31][33] which highlights the importance of sequencing white blood cells to at least the same depth as cfDNA to correctly exclude the CH mutations and avoid misinterpretation. Furthermore, our group has recently shown there are no significant differences between the VAF of CH-related and tumor-derived ctDNA detected in plasma samples [32]. The indifferences in the type of variant and VAF between CH and ctDNA mutations reinforce the difficulties to differentiate them without performing paired deep sequencing of plasma cfDNA and DNA from white blood cells.
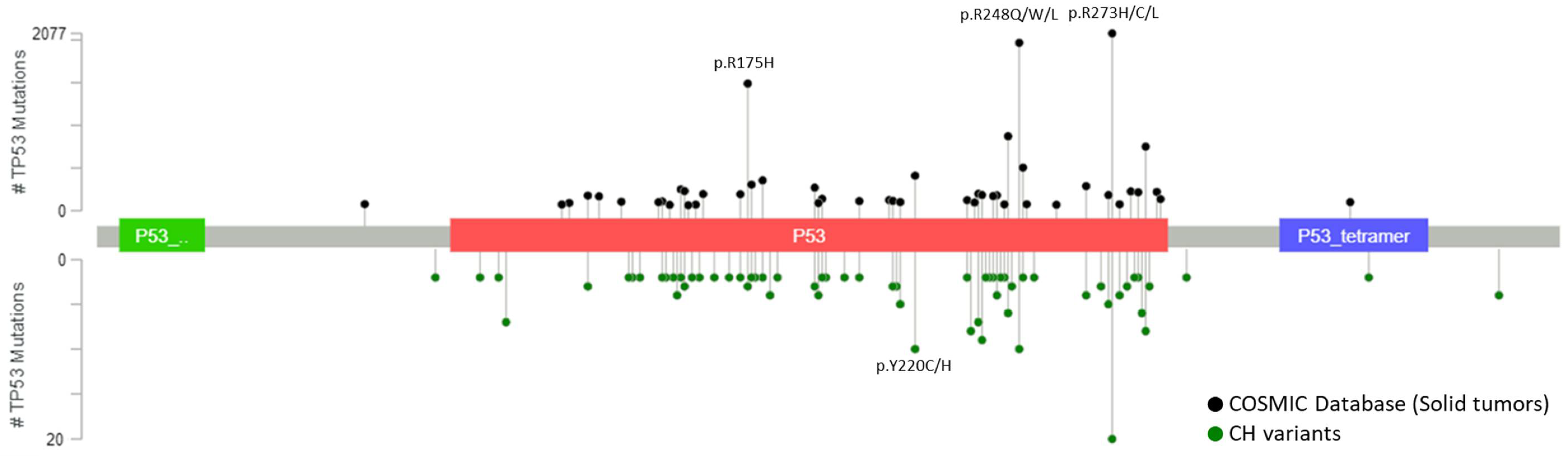
Figure 1. Most frequently detected TP53 variants from solid tumors and white blood cells. Positions and frequencies of most frequently detected TP53 mutations in solid tumors from COSMIC database (top plot) and in white blood cells from 14 published studies (bottom plot). All reported variants of TP53 from the COSMIC database were gathered and variants detected from hematopoietic and lymphoid neoplasms were filtered leaving only the variants detected from solid tumors. Variants without genomic positions documented were further removed from the analysis leaving a total of 801 CH mutations across 657 different variants. All variants that were reported more than once across the 14 studies were used for the analysis and compared with the top 85 most frequently detected TP53 variants from solid tumors. Lollipop plots were generated using the MutationMapper from cBioPortal [41][42].
Recent studies have focused on increasing the understanding of biophysical and genomic features of CH and ctDNA to assist the classification of cfDNA mutations. Several proof-of-concept studies indicated that ctDNA presents as shorter fragment size distribution than CH or non-mutated cfDNA fragments [32][35][37]. The unique shorter fragment size of ctDNA could help to identify tumor-derived cfDNA mutations. Furthermore, the majority of point mutations observed in CH are C>T transitions, which are derived from the spontaneous deamination of methylated cytosine into thymine, also consistent with the aging-related mutational signature (signature 1) [5][15][19][38][39]. Recent studies have found this age-related mutational signature to be enriched in CH cfDNA fragments and absent in ctDNA fragments [18][32]. In contrast, smoking mutational signature (signature 4) was exclusive to tumor-derived fragments and absent in CH cfDNA in NSCLC patients [32]. These observations suggest that biophysical and genomic features of cfDNA variants might be useful for distinguishing tumor-derived mutations from CH.
3.2. Detection of CH from Tumor Tissues
Tumor-informed liquid biopsy analysis has become one of the common approaches used by research groups to overcome the problem of somatic mosaicism in plasma [32][43][44][45]. It involves sequencing the tumor tissue (surgically resected or tissue biopsy) to identify and select tumor-specific mutations that are to be further monitored using plasma cfDNA for various clinical applications. The theory that underlies this approach is that all mutations detected from tumor tissues would be specific to the malignancy. However, such an approach should also be carefully considered as CH mutations may also be present in tumor tissues due to tumor-infiltrating blood cells [15][21][34]. Two large retrospective studies analyzed existing NGS data of paired tumor tissues and white blood cells from thousands of cancer patients with various solid tumors to assess the prevalence of CH mutations in tumor tissues [15][21]. A total of 14–77% of CH mutations detected in white blood cells were detected in tumor tissues. The VAF of CH mutations detected in tumor tissues ranged from 0.5 to 21% [15][21]. The limit of detection for an NGS assay is directly correlated to the sequencing depth. The VAF of CH in tumor tissues could go below 0.5% with increasing sequencing depth and their prevalence could be higher than previously reported [34]. These results highlight the importance of white blood cell sequencing even for tumor-informed analysis to exclude the possibility of CH. The clinical implications of these findings are not limited to liquid biopsy, CH mutations should also be considered when performing tumor profiling using tumor tissues to prevent incorrect identification of targetable alterations.
3.3. Clinical Impact of CH Mutations on the Interpretation of Liquid Biopsy Results
The presence of CH in cfDNA of the general population has now been well established, however, limited studies have been conducted to directly examine the impact of CH on the clinical interpretation of blood liquid biopsy results. cfDNA analysis has been suggested as a promising tool for cancer screening. As mentioned in previous studies, a large proportion of mutations detected in plasma cfDNA of healthy individuals could be originated from CH [18][31][33]. Misinterpretation of these CH-related mutations as ctDNA mutations may lead to unreliable diagnosis.
Liquid biopsy was recently approved to identify actionable alterations in specific genes that could assist in treatment selection when tumor tissue is unavailable. CH mutations should be carefully considered for these assays as recent studies have shown that a substantial number of CH variants are considered to be oncogenic and are indicated for molecular-targeted therapies [24][31][35]. In the study conducted by Razavi et al., up to 10% of the CH mutations detected in plasma were listed as oncogenic in OncoKb database and 13% of these mutations were indicated for either an approved targeted therapy or a treatment under clinical trial. Incorrect identification of actionable alteration would lead to inappropriate treatment.
The minimal invasiveness of liquid biopsy and the ability for serial sampling highlight its usefulness for the detection of minimal residual disease and monitoring of treatment response for solid tumor patients [46][47][48]. However, most of the studies conducted earlier did not exclude CH mutations in the analysis. We evaluated the impact of misclassification of CH as ctDNA on the clinical interpretation of cfDNA results in one of our recent studies [34]. In our colorectal cancer patients study cohort, 17% of the pre-operative cfDNA mutations were identified to be CH-related. The recruited patients were followed post-operatively and the identified CH mutations were recurrently detected after surgery or completion of adjuvant chemotherapy. Under unpaired sequencing of cfDNA, the consistent detection of CH could be incorrectly interpreted as the presence of residual disease after tumor resection or inappropriately inferred as disease progression or treatment ineffectiveness.
4.4. Other Potential Sources of Non-Tumoral Somatic Mutations
Results from others and our study have shown that, besides CH from white blood cells and tumor-derived mutations, there are still other cfDNA mutations detected in the plasma with an unknown origin [31][34]. Future studies are needed to investigate whether hepatocytes and endothelial cells may also contribute to cfDNA mutations detected in plasma. Furthermore, in healthy subjects, DNA from the erythroid lineage may contribute up to 30% of plasma cfDNA . It has been suggested that CH may also affect the erythroid lineage and carries unique somatic mutations that are different from white blood cells [49]. Although mature red blood cells do not have a nucleus, erythroblasts lose their nuclei and are matured into reticulocytes in the bone marrow during the enucleation step. The nuclear material of the erythroblasts gets degraded and may be released into the circulation as a form of cfDNA, therefore, it is of great interest to investigate whether cells in the erythroid lineage are another contributor to the cfDNA mutations detected in plasma.
4. Future Directions: Clinical Interpretation of CH
The rapid development and validation of ctDNA-based liquid biopsy in observational studies and clinical trials has allowed several ctDNA-guided therapies to be approved for their use in cancer patients. Furthermore, the full clinical utilities of ctDNA in oncological management are also currently being explored through large-scale interventional clinical trials [50]. However, cfDNA analysis should be further optimized to support the use of blood liquid biopsy as a routine practice with a clinically affordable cost in cancer management.
The current approach of performing paired cfDNA-white blood cells DNA sequencing to differentiate CH mutations doubles the costs involved which may reduce the cost-effectiveness of ctDNA analysis. In the study conducted by Chabon et al., the authors utilized machine learning to incorporate molecular and genomic features of CH and tumor-derived cfDNA fragments to identify the sources of cfDNA mutations [32]. Integration of biophysical, genomic, and molecular features of CH and ctDNA together with large datasets of CH mutations and tumor mutation profile may refine the machine learning model to assist the identification of ctDNA mutations.
Our understanding of the contribution of CH to the mutations detected by cfDNA analysis has improved tremendously in the past decade. However, there are still many unknowns and unanswered questions that need to be addressed to maximize the potential utilities of blood liquid biopsy. CH-related mutations detected from liquid biopsy may provide important information for assessing hematologic malignancy and CVD risks in healthy individuals. Although the association of CH and its clinical implications has been well documented, a clear VAF cut-off to determine the relative or absolute risk of developing diseases should be further investigated. Current detection of CH mutations is often unintentional; further investigation is needed to assess whether monitoring healthy individuals who are CHIP carriers would bring any clinical benefits by allowing earlier detection of hematological malignancy or CVD. Similarly, in the field of oncology, cancer patients who are CHIP carriers are more prone to develop t-MN after chemotherapy. Future studies should assess the overall clinical benefits of chemotherapy in CHIP carriers with long-term monitoring.
This entry is adapted from the peer-reviewed paper 10.3390/cancers12082277
References
- Alix-Panabières, C. Perspective: The future of liquid biopsy. Nature 2020, 579, S9.
- De Rubis, G.; Rajeev Krishnan, S.; Bebawy, M. Liquid Biopsies in Cancer Diagnosis, Monitoring, and Prognosis. Trends Pharmacol. Sci. 2019, 40, 172–186, doi:10.1016/j.tips.2019.01.006.
- Bellosillo, B.; Montagut, C. High-accuracy liquid biopsies. Nat. Med. 2019, 25, 1820–1821, doi:10.1038/s41591-019-0690-1.
- Lui, Y.Y.; Chik, K.W.; Chiu, R.W.; Ho, C.Y.; Lam, C.W.; Lo, Y.M. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin. Chem. 2002, 48, 421–427.
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498, doi:10.1056/NEJMoa1408617.
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278, doi:10.1016/j.cell.2012.06.023.
- Gillis, N.K.; Ball, M.; Zhang, Q.; Ma, Z.; Zhao, Y.; Yoder, S.J.; Balasis, M.E.; Mesa, T.E.; Sallman, D.A.; Lancet, J.E.; et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: A proof-of-concept, case-control study. Lancet Oncol. 2017, 18, 112–121, doi:10.1016/S1470-2045(16)30627-1.
- Champion, K.M.; Gilbert, J.G.; Asimakopoulos, F.A.; Hinshelwood, S.; Green, A.R. Clonal haemopoiesis in normal elderly women: Implications for the myeloproliferative disorders and myelodysplastic syndromes. Br. J. Haematol. 1997, 97, 920–926, doi:10.1046/j.1365-2141.1997.1933010.x.
- Fey, M.F.; Liechti-Gallati, S.; von Rohr, A.; Borisch, B.; Theilkas, L.; Schneider, V.; Oestreicher, M.; Nagel, S.; Ziemiecki, A.; Tobler, A. Clonality and X-inactivation patterns in hematopoietic cell populations detected by the highly informative M27 beta DNA probe. Blood 1994, 83, 931–938.
- Lee-Six, H.; Obro, N.F.; Shepherd, M.S.; Grossmann, S.; Dawson, K.; Belmonte, M.; Osborne, R.J.; Huntly, B.J.P.; Martincorena, I.; Anderson, E.; et al. Population dynamics of normal human blood inferred from somatic mutations. Nature 2018, 561, 473–478, doi:10.1038/s41586-018-0497-0.
- Steensma, D.P. Clinical consequences of clonal hematopoiesis of indeterminate potential. Blood Adv. 2018, 2, 3404–3410, doi:10.1182/bloodadvances.2018020222.
- Genovese, G.; Kahler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487, doi:10.1056/NEJMoa1409405.
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130, 742–752, doi:10.1182/blood-2017-02-769869.
- Desai, P.; Mencia-Trinchant, N.; Savenkov, O.; Simon, M.S.; Cheang, G.; Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, M.L.; Ballman, K.V.; et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 2018, 24, 1015–1023, doi:10.1038/s41591-018-0081-z.
- Abelson, S.; Collord, G.; Ng, S.W.K.; Weissbrod, O.; Mendelson Cohen, N.; Niemeyer, E.; Barda, N.; Zuzarte, P.C.; Heisler, L.; Sundaravadanam, Y.; et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018, 559, 400–404, doi:10.1038/s41586-018-0317-6.
- Gibson, C.J.; Steensma, D.P. New Insights from Studies of Clonal Hematopoiesis. Clin. Cancer Res. 2018, 24, 4633–4642, doi:10.1158/1078-0432.CCR-17-3044.
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S.; et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 2015, 518, 552–555, doi:10.1038/nature13968.
- Fernandez-Cuesta, L.; Perdomo, S.; Avogbe, P.H.; Leblay, N.; Delhomme, T.M.; Gaborieau, V.; Abedi-Ardekani, B.; Chanudet, E.; Olivier, M.; Zaridze, D.; et al. Identification of Circulating Tumor DNA for the Early Detection of Small-cell Lung Cancer. EBioMedicine 2016, 10, 117–123, doi:10.1016/j.ebiom.2016.06.032.
- Chabon, J.J.; Hamilton, E.G.; Kurtz, D.M.; Esfahani, M.S.; Moding, E.J.; Stehr, H.; Schroers-Martin, J.; Nabet, B.Y.; Chen, B.; Chaudhuri, A.A.; et al. Integrating genomic features for non-invasive early lung cancer detection. Nature 2020, 580, 245–251, doi:10.1038/s41586-020-2140-0.
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1, doi:10.1126/scisignal.2004088.
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Barhdadi, A.; Bourgoin, V.; Lepine, G.; Mollica, L.; Szuber, N.; Dube, M.P.; Busque, L. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 2017, 130, 753–762, doi:10.1182/blood-2017-04-777029.
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478, doi:10.1038/nm.3733.
- McKerrell, T.; Park, N.; Moreno, T.; Grove, C.S.; Ponstingl, H.; Stephens, J.; Crawley, C.; Craig, J.; Scott, M.A.; Hodkinson, C.; et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015, 10, 1239–1245, doi:10.1016/j.celrep.2015.02.005.
- Marass, F.; Stephens, D.; Ptashkin, R.; Zehir, A.; Berger, M.F.; Solit, D.B.; Diaz, L.A.; Tsui, D.W.Y. Fragment Size Analysis May Distinguish Clonal Hematopoiesis from Tumor-Derived Mutations in Cell-Free DNA. Clin. Chem. 2020, 66, 616–618, doi:10.1093/clinchem/hvaa026.
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131, doi:10.1001/jamaoncol.2019.0528.
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404, doi:10.1158/2159-8290.CD-12-0095.
- Wang, Y.; Li, L.; Cohen, J.D.; Kinde, I.; Ptak, J.; Popoli, M.; Schaefer, J.; Silliman, N.; Dobbyn, L.; Tie, J.; et al. Prognostic Potential of Circulating Tumor DNA Measurement in Postoperative Surveillance of Nonmetastatic Colorectal Cancer. JAMA Oncol. 2019, 5, 1118–1123, doi:10.1001/jamaoncol.2019.0512.
- Shepherd, M.S.; Li, J.; Wilson, N.K.; Oedekoven, C.A.; Li, J.; Belmonte, M.; Fink, J.; Prick, J.C.M.; Pask, D.C.; Hamilton, T.L.; et al. Single-cell approaches identify the molecular network driving malignant hematopoietic stem cell self-renewal. Blood 2018, 132, 791–803, doi:10.1182/blood-2017-12-821066.
- Coombs, C.C.; Zehir, A.; Devlin, S.M.; Kishtagari, A.; Syed, A.; Jonsson, P.; Hyman, D.M.; Solit, D.B.; Robson, M.E.; Baselga, J.; et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 2017, 21, 374–382.e4, doi:10.1016/j.stem.2017.07.010.
- Osorio, F.G.; Rosendahl Huber, A.; Oka, R.; Verheul, M.; Patel, S.H.; Hasaart, K.; de la Fonteijne, L.; Varela, I.; Camargo, F.D.; van Boxtel, R. Somatic Mutations Reveal Lineage Relationships and Age-Related Mutagenesis in Human Hematopoiesis. Cell Rep. 2018, 25, 2308–2316, doi:10.1016/j.celrep.2018.11.014.
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131, doi:10.1001/jamaoncol.2019.0528.
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404, doi:10.1158/2159-8290.CD-12-0095.
- Wang, Y.; Li, L.; Cohen, J.D.; Kinde, I.; Ptak, J.; Popoli, M.; Schaefer, J.; Silliman, N.; Dobbyn, L.; Tie, J.; et al. Prognostic Potential of Circulating Tumor DNA Measurement in Postoperative Surveillance of Nonmetastatic Colorectal Cancer. JAMA Oncol. 2019, 5, 1118–1123, doi:10.1001/jamaoncol.2019.0512.
- Dorsheimer, L.; Assmus, B.; Rasper, T.; Ortmann, C.A.; Ecke, A.; Abou-El-Ardat, K.; Schmid, T.; Brune, B.; Wagner, S.; Serve, H.; et al. Association of Mutations Contributing to Clonal Hematopoiesis With Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol. 2019, 4, 25–33, doi:10.1001/jamacardio.2018.3965.
- Gibson, C.J.; Lindsley, R.C.; Tchekmedyian, V.; Mar, B.G.; Shi, J.; Jaiswal, S.; Bosworth, A.; Francisco, L.; He, J.; Bansal, A.; et al. Clonal Hematopoiesis Associated With Adverse Outcomes After Autologous Stem-Cell Transplantation for Lymphoma. J. Clin. Oncol. 2017, 35, 1598–1605, doi:10.1200/JCO.2016.71.6712.
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121, doi:10.1056/NEJMoa1701719.
- Tie, J.; Cohen, J.D.; Wang, Y.; Christie, M.; Simons, K.; Lee, M.; Wong, R.; Kosmider, S.; Ananda, S.; McKendrick, J.; et al. Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage III Colon Cancer. JAMA Oncol. 2019, 5, 1710–1717, doi:10.1001/jamaoncol.2019.3616.
- Tie, J.; Cohen, J.D.; Wang, Y.; Christie, M.; Simons, K.; Lee, M.; Wong, R.; Kosmider, S.; Ananda, S.; McKendrick, J.; et al. Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage III Colon Cancer. JAMA Oncol. 2019, 5, 1710–1717, doi:10.1001/jamaoncol.2019.3616.
- Chen, Y.H.; Hancock, B.A.; Solzak, J.P.; Brinza, D.; Scafe, C.; Miller, K.D.; Radovich, M. Next-generation sequencing of circulating tumor DNA to predict recurrence in triple-negative breast cancer patients with residual disease after neoadjuvant chemotherapy. NPJ Breast Cancer 2017, 3, 24, doi:10.1038/s41523-017-0028-4.
- Ng, S.B.; Chua, C.; Ng, M.; Gan, A.; Poon, P.S.; Teo, M.; Fu, C.; Leow, W.Q.; Lim, K.H.; Chung, A.; et al. Individualised multiplexed circulating tumour DNA assays for monitoring of tumour presence in patients after colorectal cancer surgery. Sci. Rep. 2017, 7, 40737, doi:10.1038/srep40737.
- Scholer, L.V.; Reinert, T.; Orntoft, M.W.; Kassentoft, C.G.; Arnadottir, S.S.; Vang, S.; Nordentoft, I.; Knudsen, M.; Lamy, P.; Andreasen, D.; et al. Clinical Implications of Monitoring Circulating Tumor DNA in Patients with Colorectal Cancer. Clin. Cancer Res. 2017, 23, 5437–5445, doi:10.1158/1078-0432.CCR-17-0510.
- Mayrhofer, M.; De Laere, B.; Whitington, T.; Van Oyen, P.; Ghysel, C.; Ampe, J.; Ost, P.; Demey, W.; Hoekx, L.; Schrijvers, D.; et al. Cell-free DNA profiling of metastatic prostate cancer reveals microsatellite instability, structural rearrangements and clonal hematopoiesis. Genome Med. 2018, 10, 85, doi:10.1186/s13073-018-0595-5.
- Chen, Y.H.; Hancock, B.A.; Solzak, J.P.; Brinza, D.; Scafe, C.; Miller, K.D.; Radovich, M. Next-generation sequencing of circulating tumor DNA to predict recurrence in triple-negative breast cancer patients with residual disease after neoadjuvant chemotherapy. NPJ Breast Cancer 2017, 3, 24, doi:10.1038/s41523-017-0028-4.
- Ng, S.B.; Chua, C.; Ng, M.; Gan, A.; Poon, P.S.; Teo, M.; Fu, C.; Leow, W.Q.; Lim, K.H.; Chung, A.; et al. Individualised multiplexed circulating tumour DNA assays for monitoring of tumour presence in patients after colorectal cancer surgery. Sci. Rep. 2017, 7, 40737, doi:10.1038/srep40737.
- Scholer, L.V.; Reinert, T.; Orntoft, M.W.; Kassentoft, C.G.; Arnadottir, S.S.; Vang, S.; Nordentoft, I.; Knudsen, M.; Lamy, P.; Andreasen, D.; et al. Clinical Implications of Monitoring Circulating Tumor DNA in Patients with Colorectal Cancer. Clin. Cancer Res. 2017, 23, 5437–5445, doi:10.1158/1078-0432.CCR-17-0510.
- Sun, K. Clonal hematopoiesis: Background player in plasma cell-free DNA variants. Ann. Transl. Med. 2019, 7, S384, doi:10.21037/atm.2019.12.97.
- Cescon, D.W.; Bratman, S.V.; Chan, S.M.; Siu, L.L. Circulating tumor DNA and liquid biopsy in oncology. Nat. Cancer 2020, 1, 276–290, doi:10.1038/s43018-020-0043-5.
- Scholer, L.V.; Reinert, T.; Orntoft, M.W.; Kassentoft, C.G.; Arnadottir, S.S.; Vang, S.; Nordentoft, I.; Knudsen, M.; Lamy, P.; Andreasen, D.; et al. Clinical Implications of Monitoring Circulating Tumor DNA in Patients with Colorectal Cancer. Clin. Cancer Res. 2017, 23, 5437–5445, doi:10.1158/1078-0432.CCR-17-0510.
- Sun, K. Clonal hematopoiesis: Background player in plasma cell-free DNA variants. Ann. Transl. Med. 2019, 7, S384, doi:10.21037/atm.2019.12.97.
- Cescon, D.W.; Bratman, S.V.; Chan, S.M.; Siu, L.L. Circulating tumor DNA and liquid biopsy in oncology. Nat. Cancer 2020, 1, 276–290, doi:10.1038/s43018-020-0043-5.