Ion channels play an important role in vascular function and pathology. In this review we gave an overview of recent findings and discussed the role of TRPC and TRPV channels as major regulators of cellular remodeling and consequent vascular disorders. Here, we focused on their implication in 4 relevant vascular diseases: systemic and pulmonary artery hypertension, atherosclerosis and restenosis.
- vascular tone
- vascular disease
- vascular remodeling
Transient receptor potentials (TRPs) are non-selective cation channels that are widely expressed in vascular beds. They contribute to the Ca2+ influx evoked by a wide spectrum of chemical and physical stimuli, both in endothelial and vascular smooth muscle cells. Within the superfamily of TRP channels, different isoforms of TRPC (canonical) and TRPV (vanilloid) have emerged as important regulators of vascular tone and blood flow pressure. Additionally, several lines of evidence derived from animal models, and even from human subjects, highlighted the role of TRPC and TRPV in vascular remodeling and disease. Dysregulation in the function and/or expression of TRPC and TRPV isoforms likely regulates vascular smooth muscle cells switching from a contractile to a synthetic phenotype. This process contributes to the development and progression of vascular disorders, such as systemic and pulmonary arterial hypertension, atherosclerosis and restenosis.
- Introduction
Blood vessels are composed essentially of two interacting cell types: endothelial cells (ECs) from the tunica intima lining of the vessel wall and vascular smooth muscle cells (VSMCs) from tunica media of the vascular tube. Blood vessels are a complex network, and they differ according to the tissue to which they belong, having diverse cell expressions, structures and functions [1–3]. Blood vessels’ heterogeneity is further enhanced by pathophysiological stimuli [4], showing different characteristics and behaviors to certain diseases such as atherosclerosis [5], hypertension [6], restenosis [7] or thrombosis [8]. Nevertheless, within the variability, there are common features that define blood vessels. Inside vessels, the blood flow and pressure are coordinated by multiple mechanisms, which can be classified into extrinsic ones, including hormonal and neuronal regulation, and intrinsic ones through myogenic and metabolic regulation [9]. Local intrinsic factors are able to activate different intracellular pathways to stimulate the required physiological response. Most of the signaling cascades trigger an increase of the intracellular calcium concentration ([Ca2+]i), which, in turn, activates signaling pathways and ion channels such as the Ca2+-activated K+ channel, Ca2+-activated chloride channel and transient receptor potential (TRP) channel [10,11]. Therefore, Ca2+ acts as a second messenger in numerous primary functions of VSMCs such as vascular tone, cell proliferation, vasculogenesis or vasoactive factors release, as reviewed in [12].
The endothelium is the primary tissue responsible for the regulation of VSMCs’ contractility, vascular wall permeability, angiogenesis, triggering coagulation and fibrinolysis and the regulation of the vascular tone and vessels diameter, as reviewed in [13,14]. In all these mechanism, endothelium production of nitric oxide (NO), prostaglandin and the secretion of vasoactive agonists play a critical role [15]. In the case of VSMCs, they are normally quiescent and contractile, although they change to a proliferative and migratory state in certain conditions, such as arterial injury or inflammation [16]. This change is considered a characteristic step in the pathogenesis of multiple vascular diseases and is associated with ion channels’ plasticity and a rise in [Ca2+]i, which activate Ca2+-dependent factors of transcription [17]. Among other channels, TRP channels seem to be implicated in the [Ca2+]i enhancement during VSMCs’ proliferation and vascular remodeling [18].
This review is organized into two main sections. The first provides a brief overview of the TRP channel expression and physiological function in vascular beds. The second considers the role of TRPC and TRPV channels in vascular remodeling and disorder, such as systemic and pulmonary arterial hypertension, atherosclerosis and restenosis, as indicated in Figure 1. In this review, we will focus in particular on the roles of TRPC and TRPV, which are among the most important TRPs in vascular beds.
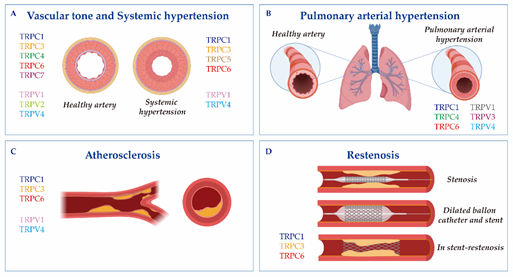
Figure 1. The cartoon indicates Transient receptor potential canonical (TRPC) and vanilloid (TRPV) isoforms involved in (A) vascular tone and systemic hypertension; (B) pulmonary arterial hypertension; (C) atherosclerosis; and (D) restenosis. Vascular responses and disorders depend either on TRP activation in the endothelium and vascular smooth muscle cells, or on critical changes in their expression.
- TRPC and TRPV Channels in Vascular Diseases
Vascular remodeling relies on molecular, cellular and structural changes in the blood vessel, which happen in response to chronic alterations in the blood flow or in response to vessel wall injury. This process is brought about by some cardiovascular diseases, like systemic or pulmonary hypertension, atherosclerosis or restenosis [57]. Ca2+ signaling and TRP channels stand as significant regulators of the VSMCs transition from a normal physiological phenotype to a dysfunctional diseased phenotype (Figure 1, Table 1).
2.1. Systemic Hypertension
Hypertension is a complex disease caused by genetic and environmental interactions characterized by a persistent increase in the vascular tone and an augmented blood pressure. The main pathological change in hypertension is vascular remodeling, which is associated with an increase in the media thickness due to VSMCs hyperplasia and/or hypertrophy [58]. Other key pathological features of hypertension are an impaired bioavailability of NO by ECs and the secretion of vasoactive agonists, which in turn modulate Ca2+ signaling in VSMCs [59]. Different isoforms of TRP channels contribute to hypertension-evoked vascular remodeling and enhanced vasoconstriction responses thanks to the alteration of [Ca2+]i and VSMCs proliferation [60,61].
TRPCs: Several studies used mouse model of essential hypertension or spontaneously hypertensive rats (SHRs) to demonstrate the significant upregulation of TRPC isoforms in aorta, mesenteric or carotid arteries [62–64]. In mesenteric arterioles from SHRs, the expression of TRPC1, C3 and C5 was augmented and correlated with an increase in norepinephrine-induced oscillations of the arterial vascular tone, known as vasomotion [64]. Antibodies against TRPC1, C3 or C5 inhibited a norepinephrine-induced Ca2+ increase and vasomotion in these resistance arteries, confirming the implication of these TRPCs in this process. Lin et al. [63] also showed that TRPC1, C3 and C6 were upregulated in the carotid artery of SHRs. Nevertheless, they demonstrated that only TRPC1 and C6 were involved in the increase of the medial thickness, lumen diameter, medial area, collagen deposition and medial VSMCs hyperplasia, typical indicators of arterial remodeling observed with age and systolic blood pressure [63]. Meanwhile, TRPC3 upregulation was found in eight-week SHRs without carotid arterial remodeling, but not in 18-week SHRs with arterial remodeling, indicating that TRPC3 may be important for hypertension development. In contrast, a previous report by Noorani et al. [65] found an increased TRPC3 and decreased TRPC1 expression at the protein levels in the carotid artery of SHRs. The authors proposed that TRPC3 overexpression in hypertension led to a greater Ca2+ and Na+ influx, depolarization and consequent activation of VDCCs, which enhanced carotid artery contractility [65]. Recently, Alvarez–Miguel et al. [62] effectively demonstrated that hypertension promoted changes in TRPC3 and C6 heteromultimeric assembly, which favored VSMCs depolarization. In accordance with these findings, an increased TRPC3 expression was detected in purified mitochondria in the vasculature of SHRs when compared to Wistar–Kyoto rats. Moreover, TRPC3-KO mice showed a reduced angiotensin II-induced reactive oxygen species (ROS) production, which suppressed vasoconstriction and decreased blood pressure [66]. TRPC3 upregulation was also observed in the mesenteric arteries and in aortic tissue of SHRs, where it was associated with exacerbated vasoconstrictions to ET-1 and angiotensin II during hypertension [67,68]. Interestingly, the upregulation of TRPC3 was also detected in monocytes from SHRs [69] and from patients with essential hypertension [61]. This overexpression correlated with an increase in the release of pro-inflammatory cytokines, such as IL-1β and TNF-α, in patients with essential hypertension when compared to normotensive control subjects [70]. In the case of TRPC6, an early study by Dietrich et al. [34] observed an elevated blood pressure and enhanced agonist-induced contractility in the aortic and cerebral arteries of TRPC6-KO mice. However, independent studies demonstrated that TRPC6 was upregulated under hypertension, using for example deoxycorticosterone acetate-salt hypertensive rats model [34] or the Milan hypertensive strain (MHS) [71]. For instance, Bae et al. [72] showed that the TRPC6 expression was enhanced in the mesenteric artery of DOCA-salt hypertensive rats, suggesting that aldosterone evoked TRPC6 upregulation, receptor-operated Ca2+ entry (ROCE) and consequently hypertension development.
Table 1. List of TRPC and TRPV isoforms involved in vascular disease. ApoE—KO, Apolipoprotein E knock-out mice; ECs, Endothelial cells; NF-κB, Nuclear factor kappa B; PAH, Pulmonary arterial hypertension; PASMCs, Pulmonary artery smooth muscle cells; ROS, Reactive oxygen species; SHRs, Spontaneously hypertensive rats; TRPC, Transient receptor potential canonical channel; TRPV, Transient receptor potential vanilloid-related channel; VSMCs, Vascular smooth muscle cells.
|
Disease |
|
Trp Channel and Mechanism |
|
Systemic hypertension |
TRPC TRPV |
Upregulation of TRPC1, C3, C5 and C6 in arteries SHRs [62–64]. They are involved in agonist-induced Ca2+ increase and vasomotion in arteries from SHRs [64]. TRPC1 and C6 are implicated in arterial remodeling in SHRs [63]. TRPC3 plays a role in hypertension development [63], ROS production and blood pressure [66]. TRPC6 is involved in aldosterone-induced Ca2+ influx and hypertension [72]. TRPV1 is downregulated in Dahl salt-sensitive rats [73]. TRPV1 activation decreases blood pressure [73,74], arterial remodeling [73], and improves nitric oxide production [75,76]. TRPV4 is implicated in VSMCs hyperpolarization and blood pressure regulation [77–81]. |
|
Pulmonary arterial hypertension |
TRPC TRPV |
Upregulation of TRPC1, C3, C5 and C6 in the human PASMCs of patients with idiopathic PAH [81–86]. Upregulation of TRPC1, C4 and C6 increases Ca2+ influx, proliferation and migration of PASMCs [82,83]. TRPC4 inactivation prevents pulmonary vascular remodeling [87]. TRPC6 overexpression promotes PASMCs’ proliferation [86,88]. Upregulation of TRPV1, V3 and V4 in the PASMCs of patients with idiopathic PAH [89–91]. TRPV1 and TRPV4 activation evokes Ca2+ influx, proliferation and contraction of PASMCs [85,89,90,92]. TRPV4 plays a role in fibroblast proliferation and the synthesis of the extracellular matrix in lungs [93]. TRPV3 participates in hypoxia-induced PASMCs’ proliferation and remodeling [91]. |
|
Atherosclerosis |
TRPC TRPV |
TRPC3 overexpression increases ECs' inflammation and macrophage infiltration [94,95]. TRPC3 deletion in macrophages reduces their presence in the atheroma plaque [96]. TRPC3 inhibition suppresses the NF-κB pathway, promotes cell viability and inhibits apoptosis in ECs.[97]. TRPC6 inhibition prevents ECs apoptosis [98]. TRPV1 activation reduces atherosclerotic lesions in the aorta of ApoE-KO mice [99], inhibits VSMCs proliferation [100] and prevents inflammation and oxidation [87,101]. TRPV4 activation prevents atherosclerosis progression [102]. |
|
Restenosis |
TRPC |
Overexpression of TRPC1 and TRPC6 correlates with Ca2+ handling in balloon-injured human internal mammary arteries [103], pig coronary arteries [104] and rat carotid arteries [105]. TRPC1 antibody prevents neointima progression in human veins [106]. Stent implantation in the aorta promotes TRPC3 upregulation [107]. |
TRPVs: Studies using TRPV1, V4-KO mice and pharmacological compounds that selectively target both channels, suggested that they regulate blood pressure and play a protective role against hypertension [108,109]. In Dahl salt-sensitive rats, TRPV1 inhibition, using capsazepine, enhanced the blood pressure remarkably; meanwhile, its activation with capsaicin decreased the mean arterial pressure in a dose-dependent manner [74]. Zhang et al. [73] also showed that the expression of TRPV1 in mesenteric arteries and the kidney was downregulated in this rat model of hypertension. Moreover, TRPV1 activation by capsaicin inhibited hypertension-induced VSMCs phenotypic switching, reduced the intracranial arteriole remodeling [73] and improved the endothelial production of NO in rats, which prevented hypertension [75,76]. Marshall et al. [110] further demonstrated that the blood pressure of wild-type mice fed with a high-fat diet was higher than TRPV1-KO mice’s blood pressure. Regarding the TRPV4’s role in hypertension, it was mainly related to its vasodilatory actions through the endothelium [77]. However, it has also been reported that VSMCs hyperpolarization contributed significantly in mediating TRPV4-dependent vasodilation [78]. The administration of L-NAME to inhibit nitric oxide synthase (NOS) promoted an increase of blood pressure in TRPV4-KO mice, indicating that TRPV4 mediated a vasodilatory compensatory mechanism to regulate blood pressure [79]. Other studies have demonstrated that the activation of TRPV4 with 4a-PDD reduced the basal and elevated the blood pressure in normal and high salt intake rats, suggesting that TRPV4 activation and upregulation may constitute a counter-regulatory mechanism to prevent salt-induced increases in blood pressure in Dhal-resistant rats [80]. Interestingly, Diaz–Otero et al. [81] demonstrated that the activation of the mineralocorticoid receptor impaired TRPV4-mediated relaxation in parenchymal arterioles and reduced cognitive function during hypertension. This study showed that mRNA of TRPV4 was reduced in cerebral arteries in angiotensin II-induced hypertensive mice and was recovered by mineralocorticoid receptor activation.
Therefore, there is a general consensus regarding the deleterious role of different isoforms of TRPC in systemic hypertension and their implication in arterial remodeling and in greater responses to vasoactive agonists. By contrast, TRPV1 and V4 seem to play a protective role against hypertension (Table 1).
2.2. Pulmonary Arterial Hypertension
Pulmonary arterial hypertension (PAH) is a hemodynamic disorder caused by pulmonary vasculature remodeling, vasoconstriction and thrombosis, responsible for a progressive elevation of the pulmonary arterial pressure that leads to right heart failure, and even death [93,111]. Chronic hypoxia is the main cause of pulmonary artery hypertension and is contributed by pulmonary media hypertrophy caused by the proliferation of pulmonary artery smooth muscle cells (PASMCs), which narrows the intraluminal diameter and increases resistance to blood flow [28,112].
TRPCs: TRPC channels are considered an alternative and/or additional Ca2+ influx pathway in PASMCs and pulmonary ECs (Figure 1B). As in other VSMCs, TRPC1 and STIM1 functionally associate in order to mediate store-operated Ca2+ entry (SOCE) in PASMCs [113]. Previous studies showed that TRPC1, C3 and C6 were expressed at the mRNA and protein levels in primary human PASMCs. Moreover, the chronic exposure to hypoxia of intralobar pulmonary arteries increased the expression of TRPC1 and C6, accompanied by enhanced SOCE and ROCE [113,114]. Similarly, bone morphogenetic protein 4 induced a SOCE increase, proliferation and migration of human PASMCs through TRPC1, C4 and C6 upregulation [82,83]. Interestingly, Alzoubi et al. [115] demonstrated that TRPC4 inactivation in rats decreased the acetylcholine-induced Ca2+ increase in PASMCs and reduced the severity of the occlusive pulmonary arteriopathy in a rat model of PAH exposed to Sugen 5416/hypoxia/normoxia (Su/Hx/Nx). This fact shows that the loss of TRPC4 may prevent pulmonary vascular remodeling. Moreover, Yu et al. [116] determined that TRPC6 was highly expressed in the PASMCs of patients with idiopathic pulmonary arterial hypertension (iPAH), where it was involved in PASMCs proliferation. In accordance with this data, a unique genetic variant in the promoter of the gene TRPC6 was discovered in iPAH patients [86]. Therefore, TRPC6 was suggested as a therapeutic target for PAH. Actually, Bosentan, an endothelin receptor inhibitor used clinically for iPAH patients, decreased PASMCs’ growth and proliferation through TRPC6 downregulation [88]. Furthermore, Sildenafil, another drug used widely to treat PAH, efficiently decreased TRPC1 and TRPC6 expression in the distal pulmonary arteries of chronically hypoxic rats and inhibited the chronic hypoxia-induced increase in basal [Ca2+]i and SOCE in PASMCs [84,85].
TRPVs: Independent studies suggested that TRPV1, V3 and V4 also play a role in PASMCs’ proliferation and PAH. TRPV1 activation increased [Ca2+]i in PASMCs, which enhanced vascular contraction, proliferation and migration through NFAT and CREB activation [92]. The incubation of PASMCs with hypoxia activated TRPV1 and V4, which increased PASMCs’ migration and cytoskeleton reorganization [117]. Similarly, PASMCs isolated from iPAH patients presented an overexpression of TRPV1 and V4 when compared with healthy subjects [89,90]. Furthermore, TRPV4 knockdown with siRNA significantly attenuated the shear stress-induced increase of [Ca2+]i in iPAH-PASMCs [89], confirming the role of TRPV4 as a mechanosensitive channel sensible to flow shear stress. Moreover, an increased Ca2+ entry through TRPV4 contributed to an enhanced PASMCs contraction, migration and proliferation triggered by chronic hypoxia [118]. More recently, TRPV4 was studied in pulmonary arterial adventitial fibroblasts using TRPV4-KO mice, siRNA and the pharmacologic inhibition of TRPV4 [93]. This study revealed that TRPV4 played an important role in the proliferation, migration and synthesis of the extracellular matrix, hallmarks of the pathogenesis of PAH [93]. On the other hand, TRPV3’s role in PAH was examined by Zhang et al. [91], who showed that TRPV3 was upregulated in the pulmonary vessels of PAH humans when compared to control subjects. They further demonstrated that TRPV3 participated in the hypoxia-induced proliferation of PASMCs and pulmonary vascular remodeling through the PI3K/AKT pathway [91].
In summary, PAH is associated with the upregulation of TRPC1/C3/C4/C6 and TRPV1/V3/V4 (Table 1), which play an important role in the increase of Ca2+ influx in PASMCs and in their proliferation and migration, which are critical steps for pulmonary artery remodeling.
2.3. Atherosclerosis
Atherosclerosis is a chronic inflammatory disease characterized by ECs and VSMCs proliferation and migration. During this pathogenesis, inflammatory cells, mainly monocytes and neutrophils, are recruited to the vascular wall (the site of inflammation), suffering a pathological growth that leads to atheroma plaque formation [119]. Subsequently, in the atheroma plaque, macrophages produce inflammatory cytokines and engulf lipids in an attempt to minimize the pathological accumulation of cholesterol and other lipids. In the past two decades, TRP channels have been implicated in both aspects of the disease: the proliferation of cells in the vascular wall and the pathophysiology of macrophages (Figure 1C and Table 1) [120,121].
TRPCs: Some studies reported that the reactivity of ECs to ET-1 was increased with hypercholesterolemia, considered a potent trigger in atherogenesis [122]. Bergdahl et al. [123] and Ingueneau et al. [124] demonstrated that cholesterol influenced the vascular reactivity to ET-1, affecting the caveolar localization of TRPC1. Similarly, the specific deletion of TRPC3 from bone marrow cells reduced macrophages’ presence in the atheroma plaque [125] and the specific suppression of this channel in macrophages reduced necrosis inside the atheroma plaque [96]. In contrast, the overexpression of TRPC3 in the endothelium exacerbated endothelial inflammation and macrophage infiltration, resulting in an increased burden of advanced aortic atherosclerosis [94,95]. Interestingly, a recent study by Min et al. [97] used a high-fat diet-fed apolipoprotein E knock-out (ApoE--KO) mice model of atherosclerosis to demonstrate that microRNA-26a overexpression, through TRPC3 inhibition, suppressed inflammatory responses and the NF-κB pathway, promoting cell viability and inhibiting apoptosis in oxidized low-density lipoprotein (ox-LDL)-stimulated ECs. A previous study by Zhang et al. [98] also demonstrated that this microRNA-26a was downregulated in the aortic intima of ApoE-KO mice, where it specifically inhibited TRPC6 expression and ECs apoptosis, indicating that TRPC3 and C6 suppression may prevent atherosclerosis progression.
TRPVs: Compelling evidence points to the fact that TRPV1 and V4 play a role in atherosclerosis. The long-term activation of TRPV1 by capsaicin significantly reduced lipid storage and atherosclerotic lesions in the aortic sinus and in the thoracoabdominal aorta of ApoE-KO mice, but not of ApoE and TRPV1 double KO mice [99]. TRPV1 activation also inhibited VSMCs proliferation in an atherosclerosis context [100] and prevented inflammation and oxidation [87,101]. Moreover, the protein level of TRPV1 was markedly higher in the aorta of ApoE-KO than in wild-type mice and was upregulated in ox-LDL-treated bone-marrow-derived macrophages. Therefore, the role of TRPV1 was linked to the lipid metabolism and inflammatory responses of macrophage-foam cells [126]. Similarly, Goswami et al. [102] demonstrated that TRPV4 genetic ablation or its pharmacologic inhibition blocked ox-LDL-induced macrophage foam cell formation and prevented pathophysiological matrix stiffness, indicating that TRPV4 activation might prevent atherosclerosis progression.
Altogether, these studies indicate that TRPC3/C6 and TRPV1/V4 in particular are involved in different aspects of atherosclerosis progression, while more data are needed to confirm the role of other TRPC and TRPV isoforms.
2.4. Restenosis
Restenosis is the recurrence of stenosis in vascular blood vessels that occurs mainly after angioplasty [127]. Vascular injury is characterized by an endothelial denudation that leads an inflammatory response; this results in the onset of several proliferative processes, including VMSCs switching from contractile to synthetic phenotypes, which contributes to the development and progression of restenosis [128,129]. This process is regulated by changes in Ca2+ signaling, ion channels and transcription factors activation [17,130–132]. However, only few reports have addressed the role of TRP channels in restenosis and most of them have focused on TRPC; meanwhile, there is no data regarding TRPV implication in restenosis (Figure 1D and Table 1). Bergdahl et al. [103] showed that the balloon-induced dilatation of human internal mammary arteries enhanced the plasticity of TRPC expression. Specifically, the authors observed a significant increase in the expression of TRPC1 and TRPC6 at the mRNA level, which correlated with cellular Ca2+ handling. This finding was supported by an experiment done in pig coronary arteries [104] and in human vein samples, in which the upregulation of TRPC1 was also observed after angioplasty [106]. Interestingly, TRPC1 antibodies prevented neointima progression in human vein samples cultured in vitro [106]. Recently, Jia et al. effectively showed that TRPC1 interacted with Orai1 and Homer to promote neointima formation and stenosis in balloon-injured rat carotid arteries [105]. Moreover, Koenig et al. [107] demonstrated, using a novel ex vivo organ culture model based on stent implantation in aortic constructs, that Pyr3 inhibited the stent-induced upregulation of TRPC3 expression and the proliferation of human coronary smooth muscle cells, a particular parameter of stent-induced vascular remodeling. Therefore, TRPC1,C3, and C6 seem to play a role in the restenosis of different arteries; meanwhile, information regarding the role of TRPV is lacking.
- Conclusions
Vascular remodeling and consequent disorders are highly prevalent diseases that cause significant morbidity and mortality. Increasing number of studies have shed light on the contribution of TRPC and TRPV to ECs’ and VSMCs’ physiological and pathological functions. Studies with animal models of common vascular diseases, like systemic and pulmonary hypertension, atherosclerosis and restenosis, which use KO mice of different TRP isoforms, suggest the translational potential role of TRPC and TRPV channels. Previous cutting-edge studies have suggested that these channels might be major regulators of cellular remodeling through their ability to modulate transcriptional programs that drive cell proliferation and migration, and even cytokine secretion [18,70,92]. As reviewed above, there is general agreement that most isoforms of TRPCs are implicated in cellular remodeling, a critical step for vascular disease progression, which occurs in systemic and pulmonary hypertension, atherosclerosis and restenosis. Meanwhile, TRPV1, V3 or V4 likely play a protective role in systemic hypertension and atherosclerosis, since they participate in NO release via endothelium and metabolism lipid regulation [87,101,108,109]. In contrast, TRPV1 and V4 activation seem critical for hypoxia-induced Ca2+ increase and further PASMCs proliferation and migration [92,117]. Nevertheless, future studies are urgently needed to address the downstream targets and mechanisms modulated by these channels, as well as the upstream regulators leading to changes in their expression and function during disease.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21176125