The innate and adaptive immunities have been documented to participate in the pathogenesis of nephrotoxic acute kidney injury (AKI); however, the mechanisms controlling these processes have yet to be established. In our cisplatin-induced AKI mouse model, we show pathological damage to the kidneys, with the classical markers elevated, consistent with the response to cisplatin treatment. Through assessments of the components of the immune system, both locally and globally, we demonstrate that the immune microenvironment of injured kidneys was associated with an increased infiltration of CD4+ T cells and macrophages concomitant with decreased Treg cell populations. Our cell-based assays and animal studies further show that cisplatin exposure downregulated the protein levels of programmed death-ligand 1 (PD-L1), an immune checkpoint protein, in primary renal proximal tubular epithelial cells, and that these inhibitions were dose-dependent. After orthotopic delivery of PD-L1 gene into the kidneys, cisplatin-exposed mice displayed lower levels of both serum urea nitrogen and creatinine upon PD-L1 expression. Our data suggest a renoprotective effect of the immune checkpoint protein, and thereby provide a novel therapeutic strategy for cisplatin-induced AKI.
1. Introduction
Acute kidney injury (AKI) is a clinical problem, defined by a rapid decline in renal function occurring within a short-term time span of a few hours to a few days. It is marked by tubular epithelial damage, with resultant damage and the loss of kidney function, causing an accumulation of waste products, and a loss of electrolytes and the acid-base balance [
1]. AKI is a significant clinical problem, with 4 million hospitalization cases in 2014, and this number continues to grow year every year. This troubling increase indicates a growing health concern and underscores the importance of understanding the pathogenic mechanisms of this problem [
2]. Of the many wide-ranging and often multifactorial causes of AKI, drug nephrotoxicity represents a significant portion of cases in the clinical setting, accounting for between 8–60% of AKI cases, depending on the cohort [
3]. A large multicenter study indicated that up to 19% of AKI cases could be attributed to drug toxicities [
4], further supporting this observation. This could primarily be attributed to the function of the kidney as a filter; thus, it is a major target site that is exposed to the toxic effects of therapeutic drugs and their metabolites as the kidney performs its normal function in filtering the circulating blood.
Cisplatin (cis-diammine-dichloro-platinum II), and other platinum chemotherapy drugs, are among the most widely used chemotherapeutic drugs for a diverse range of solid and liquid tumors [
5]. Although effective, the usage of cisplatin and other platinum-based drugs has a broad range of significant side effects, including nephrotoxicity. Up to one-third of patients experience some degree of nephrotoxicity after just one dose of cisplatin [
6]. Thus, the evaluation of the mechanisms of damage induced by cisplatin exposure, and the development of strategies to mitigate this damage are warranted to improve patient outcomes and to potentially expand the utility of cisplatin through a reduction in the nephrotoxic side effects.
One potential approach is to target the inflammatory processes evoked in response to chemotherapeutic treatment. Most AKI has been demonstrated over recent years to be a systemic disease, with inflammation playing a critical part in the disease process [
7]. After the initial injury of the tubular epithelium, inflammatory cells are recruited to the site of the injury. Virtually all immune cells are involved in the process, with both adaptive and innate immunities shown to contribute to the pathogenesis of AKI [
7,
8,
9]. Given the importance of the immune system in regulating the response to kidney injury in response to toxic agents and the prevalence of cisplatin in the standard of care in cancer, it is of distinct interest to evaluate how the immune system modulates the damage associated with cisplatin-induced AKI.
Essentially all of the components of the immune system have been implicated as playing roles of variable importances and effects in nephrotoxic AKI, but the mechanisms controlling these processes are still not completely understood. In this study, we show the immune microenvironment of cisplatin-injured kidneys and demonstrate the therapeutic potential of genetically engineered kidneys with PD-L1 expression in mitigating cisplatin-mediated kidney damage.
2. Cisplatin Induces Acute Kidney Injury
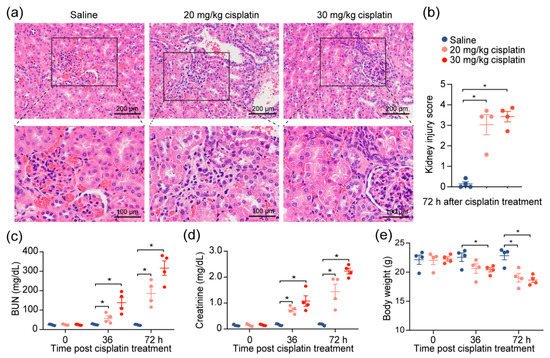
Given the observations of kidney injury in response to cisplatin, we investigated the effects of cisplatin on renal function by modifying a previously established cisplatin-induced AKI mouse model [
10]. After the intraperitoneal administration of 20 or 30 mg/kg of cisplatin, we confirmed severe renal damage in mice injected with a single dose of cisplatin, as indicated by the distortion of the tubules and tubular cell necrosis (
Figure 1a,b). We also noted an expected increase in the level of serum blood urea nitrogen (BUN) and creatinine, which increased with escalating doses (
Figure 1c,d), reflecting the renal damage caused by cisplatin. To account for the possibility that diet and muscle mass content could influence the serum creatinine and BUN [
11], we evaluated the change in the body weights of the mice throughout the duration of the experiment. We found that, despite weight reduction in cisplatin-treated mice, there was no correlation between creatinine levels and weight loss (
Figure 1e). The data demonstrate that chemotherapeutic drug cisplatin decreases kidney function.
Figure 1. Cisplatin induces acute kidney injury. Mice were intraperitoneally injected with one dose of cisplatin, at 20 mg/kg or 30 mg/kg, for 72 h. Body weight, levels of BUN, and creatinine in serum were determined at 36 and 72 h. At 72 h, spleens and kidneys from mice were collected for pathology analysis and immunophenotyping, respectively. (a) Representative images, and (b) tubular injury quantification of H&E staining in kidney specimens from mice treated with 30 mg/kg cisplatin for 72 h (n = 4). (c–e): levels of serum BUN (c), creatinine (d), and body weight (e) (n = 4. * p < 0.05).
3. AKI Mice Exhibit an Imbalanced Immune Response
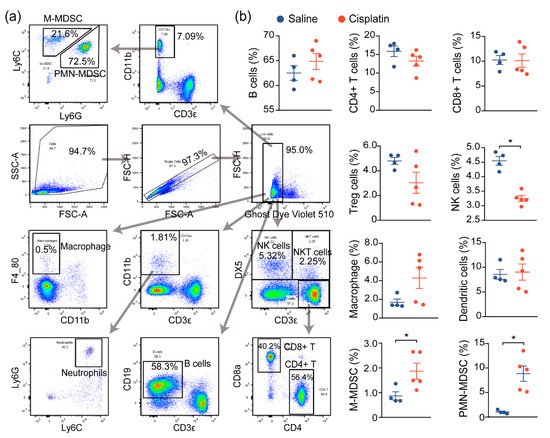
Prior reports have demonstrated that immune cell infiltration and a proinflammatory immune response contribute to the damage and subsequent renal dysfunction during AKI. In agreement with prior reports, we noted increased immune cell infiltration in cisplatin-exposed kidneys, compared to nonexposed kidneys, and we next assessed the changes in the immune profile and functionality of these cells, both locally and globally. We first evaluated the global inflammatory status by analyzing the immune cells in the spleen, a critical immune organ involved in local and systemic inflammation [
12]. Flow cytometry analysis demonstrated a significant change in the populations of monocytic and polymorphonuclear myeloid-derived suppressor cells (M-MDSC and PMN-MDSC), as well as natural killer (NK) cells, although no significant changes on the B cells, T cells (CD4+, CD8+, Treg), macrophage, or DCs were observed between saline- and cisplatin-treated mice. This suggests that the immune response may be dysregulated in response to cisplatin exposure (
Figure 2a,b).
Figure 2. AKI mice exhibit an imbalanced immune response. (a) Gating strategy for immune cell profiling of mice in Figure 1. Single cell suspension of immune cells from spleens of mice were prepared and labeled with the indicated antibodies and dyes. Data were acquired by flow cytometry and analyzed using Flowjo. (b) Analysis of splenic immune cells in mice administered with saline (n = 4), or 30 mg/kg cisplatin (n = 5, * p < 0.05).
4. Immune Characterization and Profiles of AKI
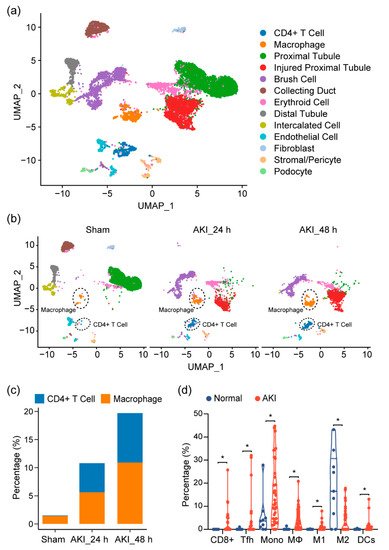
Given our observations, as well as previous reports showing that the immune profile is altered in cisplatin-induced AKI, we sought to look at the shifts in the cell population on the single-cell level in order to provide a more comprehensive assessment of the immune response in AKI and to further elucidate the cell types at play in this process. At the time of writing, single-cell data for cisplatin-induced AKI did not exist. Thus, we turned to single-cell RNA sequencing (scRNA-seq) data on mouse kidneys for ischemia/reperfusion (IR)-induced AKI, a model that resembles drug-induced AKI and shares many common aspects in the immune response. After quality control, 11,682 cells passed filtration and were subjected to clustering and dimensionality reduction with Uniform Manifold Approximation and Projection (UMAP). Clusters were further annotated by directly comparing their transcriptional profile with known cell-type-specific markers. We identified 13 cell populations, including proximal tubular cells, injured proximal tubular cells, etc. (Figure 3a). In the immune compartment, we observed elevations of the CD4+ T cells and macrophages during AKI progression (Figure 3b,c).
Figure 3. Immune characterization and profiles of AKI. (a) UMAP clustering of cell types in kidneys, from normal and AKI mice, by analysis of single-cell RNA-sequencing data (GSE139506). (b) Cell-type distribution in kidneys, from normal and AKI mice, 24 h and 48 h after induction of AKI. (c) Quantification of CD4+ T cells and macrophage cells, as percentage of total cell populations, in kidneys from normal and AKI mice. (d) Estimated proportion of the immune subpopulations in human kidney biopsy samples (GSE139061) using CIBERSORT analysis. Data are presented as mean ± SEM. AKI (n = 39) and normal (n = 9), * p < 0.05.
Next, to see if this could be observed in human tissue, we analyzed RNA-seq data of kidney biopsy tissue in dataset GSE139061. Through cell-type identification, by estimating the relative subsets of RNA transcripts through CIBERSORT, we noted that T cells, as well as macrophage cells, were the predominantly elevated immune cell populations in AKI, confirming our findings in mouse tissue (Figure 3d). In particular, we noted proinflammatory cell types (CD8+ cells, Tfh, M1, and DC cells). These elevations were also mirrored by a decrease in the M2 macrophage population, suggesting a more inflammatory environment with a decrease in reparative/anti-inflammatory cell types.
5. PD-L1 Dysregulation in Renal Epithelium upon Kidney Injury
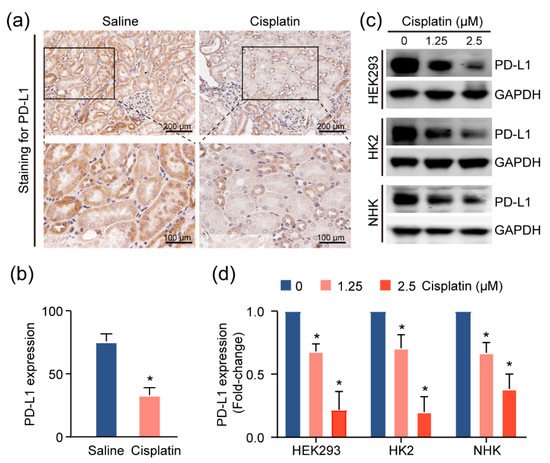
Given the increase in the T-cell population in injured kidneys, we suspected that these cells contribute to the inflammatory microenvironment in the kidney. As the activities of these T cells are associated with the expression of the immune checkpoint molecules, particularly programmed death ligand 1 (PD-L1) [
13], we utilized IHC assays to assess the levels of this immune checkpoint protein in the CAKI model mouse tissue. We observed more intense PD-L1 staining in normal tubular epithelial cells, as compared to cisplatin-exposed renal mouse epithelium (
Figure 4a,b). Consistently, treatment with cisplatin reduced PD-L1 expression in HEK293 cells, and two primary human kidney epithelial cell lines, HK2 and NHK (
Figure 4c,d), suggesting the possibility that the inflamed kidney microenvironment is attributable to cisplatin-induced PD-L1 downregulation in the renal epithelium.
Figure 4. PD-L1 dysregulation in renal epithelium upon kidney injury. (a) Representative images for PD-L1 expression levels and quantification, (b) of PD-L1, assessed by immunohistochemical (IHC) staining in cisplatin-exposed kidneys (n = 4) using anti-PD-L1 monoclonal antibody. Positive staining is quantified (mean ± SD, * p < 0.05 versus saline group). (c) Western blot analysis of kidney cell lines treated with escalating doses of cisplatin and quantification, (d) for PD-L1 levels in cells upon treatment with cisplatin for 24 h. Normalized expression levels of PD-L1 from three separate experiments are shown (* p < 0.05).
6. Targeting PD-L1 Protects against AKI
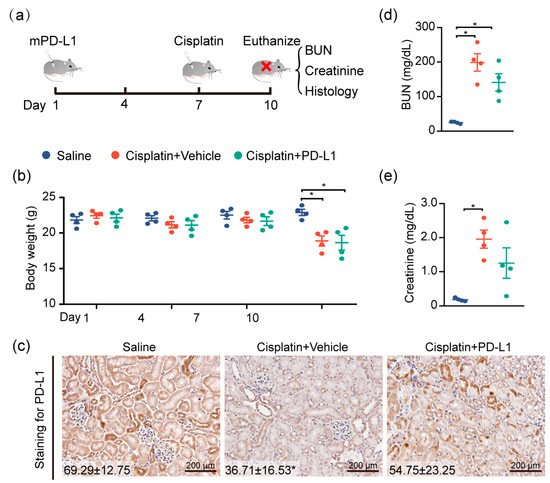
To test the renoprotective effect of PD-L1 expression, the mouse kidney was genetically engineered by utilizing the lentivirus-mediated overexpression of mouse PD-L1 (mPD-L1) prior to cisplatin exposure. Figure 5a illustrates the protocol for cisplatin administered in this study. IHC staining confirmed that kidneys receiving mPD-L1 lentivirus participles rescued the expression level of PD-L1 in tubular epithelial cells after 72 h of cisplatin exposure (Figure 5c), whereas an approximately 2-fold decrease in the PD-L1 level still occurred in the cisplatin-alone group. Surprisingly, the overexpression of mouse PD-L1 decreased the cisplatin-induced serum BUN and creatinine (Figure 5d,e), while no significant difference was noted in the weight loss between the two cisplatin-treated groups (Figure 5b). Altogether, our results show, for the first time, a critical role for PD-L1 expression in the renal epithelium in the regulation of the kidney immune microenvironment and the improvement of kidney function (Figure 6).
Figure 5. Targeting PD-L1 protects against AKI. PD-L1-containing lentiviruses were delivered into the kidneys of mice through subcapsular injection. Seven days after injection, mice were intraperitoneally treated with 30 mg/kg cisplatin for 72 h, and body weights and blood sera were collected at the end of the study, at 10 days. (a) Study timeline. (b) Body weights of mice throughout the duration of the experiment. (c) Representative images of IHC staining using anti-PD-L1 antibody (n = 4; mean ± SD). (d) The levels of serum BUN and (e) creatinine were determined (n = 4; * p < 0.05).
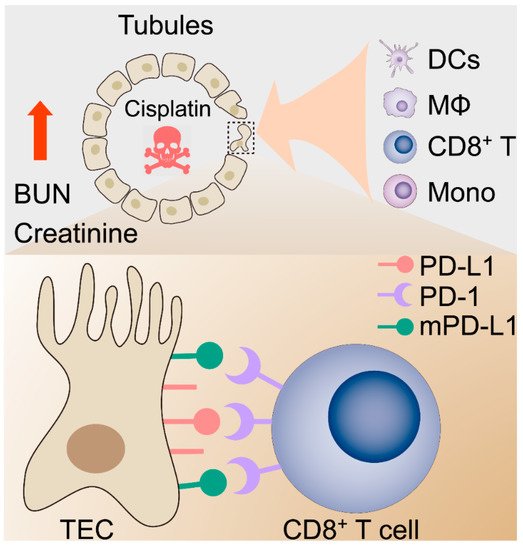
Figure 6. Proposed hypothetical model for the contribution of the kidney immune microenvironment and the PD-1/PD-L1 in nephrotoxic AKI.
This entry is adapted from the peer-reviewed paper 10.3390/ijms222413304