1. Introduction
Robert Koch identified the etiological agent of tuberculosis (TB) as
Mycobacterium tuberculosis (Mtb) [
1]. TB generates a lot of concerns as a contagious disease that poses a high risk to public health globally. Despite the available anti-tubercular drugs introduced over the years, TB remains one of the leading causes of death globally [
2]. According to the World Health Organization (WHO), it is the most common infection caused by a single bacterium. About 10 million people were diagnosed with TB in 2017, and 558,000 of them showed resistance to the most effective first-line medication, rifampicin. According to another WHO survey, an estimated 1.5 million deaths occurred in 2018 [
3]. It infects about a third of the world’s population and kills approximately 1.7–1.8 million people per year, demonstrating the failure to find new antibiotics to conquer this deadly disease [
4]. Therefore, antimicrobial compounds that are effective against Mtb are desperately required to tackle this global epidemic, worsened by resistance to medication, long-time treatment schedule, and co-infection, especially with Human Immunodeficiency Virus (HIV). In more than 40 years, no new antibiotic to treat TB has been created [
4,
5].
Recently, phenotypic screening efforts using commercial vendor libraries evolved toward identifying compounds that inhibit Mtb development [
6,
7,
8]. This intervention gives a ray of hope in the search for new therapeutics against Mtb. The urgency to end the Mtb epidemic requires improvement in diagnostic tools and the efficacy of therapeutics used in treating TB in diagnosed patients. This intervention reduces the treatment regimens usually required with strict compliance to ensure effective treatment. Rapid and cheap diagnostic test kits that can be readily accessible to the public aids early diagnosis, while drugs with multiple targets go a long way to improve the outcome of treatment [
9]. There is urgent attention to deliver new potential active antimicrobial agents to scale down the resistant TB strains. Many strategies and efforts have been adopted, which involved the structure-based design of inhibitors for a single target pathogen through computational methods [
10,
11,
12,
13].
Target drug discovery begins with identifying and studying enzymes or proteins necessary for the growth and development of the pathogen. Researchers then screen these proteins against some chemicals or compounds in libraries for potency and inhibitory effect leading to drug candidate identification using computer software after learning the accurate details of the target and lead molecule. This procedure could help pharmaceutical firms, agencies, and research labs avoid following the “false” clues. In contrast to the traditional drug discovery approach, which is time-consuming, expensive, and laborious, a new understanding of the quantitative relationship between structure and biological activity leads to the emergence of computer-aided drug design (CADD) applications in search of new therapeutics against TB. Table 1 shows the advantages of the computer-aided method of designing drugs over the traditional method.
Table 1. Comparison of the traditional method of drug development with CADD (computer-aided drug design).
| The Traditional Method of Drug Development |
CADD |
| It involves more trial-and-error processes |
It is more logical |
| It involves blind screening |
It is specific and mostly target-based |
| It is a more expensive approach to drug development |
It minimizes the cost of drug development |
| It is a relatively more laborious and time-consuming approach |
It reduces the duration required in the development of new drugs |
| It involves sequential steps |
It entails steps that are not only sequential but are also parallel and straightforward. |
| It involves separate interdisciplinary drug development with more difficult processes |
It coordinates interdisciplinary drug development with easier processes. |
The rapid advances in high-throughput screening (HTS) technologies and computational chemistry created an atmosphere that allows vast libraries of compounds to be screened and synthesized in a short period, speeding up the drug development process [
14]. CADD involves storage, management, analysis, and modeling of potential therapeutic compounds. It refers to computational methods and techniques for storing, handling, analyzing, and modeling chemical compounds. It includes computer programs for designing compounds, tools for systematically evaluating possible lead candidates, and the development of digital libraries for researching chemical interactions between molecules, among other topics [
15]. Advances in drug discovery involve using computational analysis to identify and validate vulnerable targets, which leads to the emergence of new therapeutics; they are also used in preclinical trials, drastically altering the drug development pipeline. Computational techniques can cut drug production costs by up to 50% [
16,
17,
18]. On average, it takes 10–15 years and $500–800 million to bring a drug to market, with lead analogue synthesis and testing accounting for a significant portion of that cost. As a result, using computational methods during optimization drastically reduces the expenses on drug development, as there are computational models that can screen thousands of compounds before synthesis and in vitro testing.
New therapeutics against TB emerged from HTS techniques and other related software development. There has also been an increase in biological and chemical data available on Mtb to facilitate new target identification. Furthermore, improvements in data storage capacity, supercomputing ability, and parallel processing encouraged the adoption of CADD as an integral component of TB pharmaceutical research. CADD made drug discovery all-encompassing, including different fields. Computational tools of CADD made it possible to ascribe more than 5000 macromolecular structures in the Protein Data Bank (PDB) to Mtb [
19,
20]. This repository provides a fertile ground for discovering new compounds as potent drug molecules to combat TB [
1,
19].
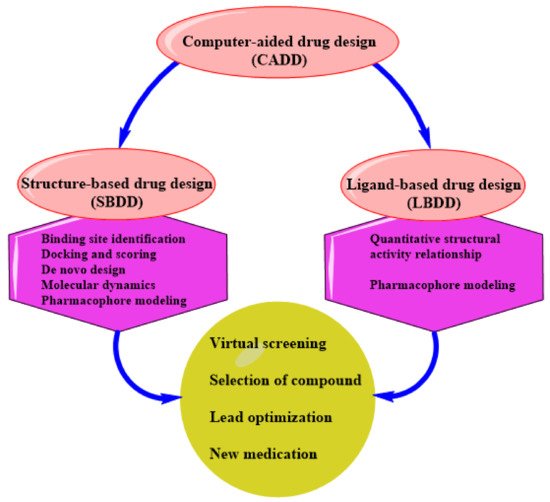
CADD can be structure-based drug design (SBDD) or ligand-based drug design (LBDD). These are the two most popular approaches to drug discovery (
Figure 1). Currently, no single method can meet all the necessities of drug discovery and production. As a result, several computational methods are used widely and effectively in combinatorial and systemic approaches [
1]. This review examines the evolution of TB tolerance, current drug management, and the development and adoption of new compounds as anti-tubercular therapeutics.
Figure 1. An illustration of CADD.
2. Status of Computational-Aided Drug Design and Discovery in TB
The drug discovery process for novel anti-tubercular therapies has evolved throughout the years due to the accumulation of biological and chemical data, the identification of numerous validated targets, and the advancement of high-throughput screening methods and software algorithm development. Aside from that, advances in data storage capacity, supercomputing power, and parallel processing allowed computer-aided drug design (CADD) to become an integrated component of TB drug design and discovery research during the last several years. As computing power continues to grow, it may soon be possible to conduct extensive exploration of the vast chemical space, which is estimated to contain about 1060 organic molecules below 500 Da, to identify potential therapeutic attractive moieties [
124] for effective Tb treatment.
Furthermore, the massive protein structural data, which includes more than 180,000 macromolecular structures available in the PDB (
www.rcsb.org accessed on 8 November 2021) and other protein repositories, gave the computational SBDD (
Figure 2) concept an impetus. The pulled structures allow identification of critical receptor catalytic and allosteric sites, molecular nature, and crucial features for in silico SBDD research. There has been much focus on TB with the countless ongoing drug discovery research, with several thousand published CADD studies. Although this is the case, Ekins et al. [
125] identified gaps in the application of computational methods in TB research, resulting in a slow stream of candidates’ drugs entering the TB drug pipelines, despite the evident need and immediacy for an effective treatment against this infection. Therefore, there is a need for more rigorous efforts to develop TB drugs leveraging the benefits provided by computational techniques.
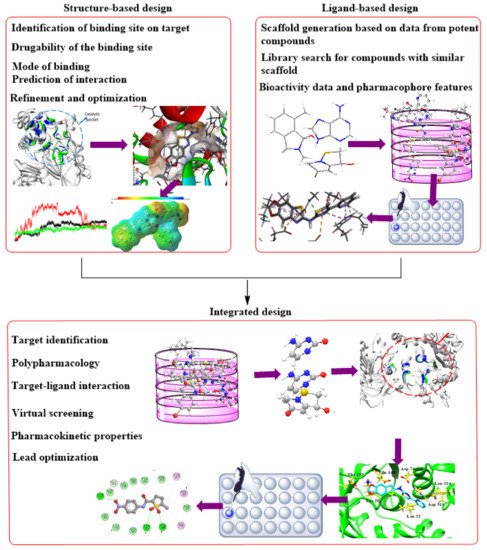
Figure 2. Complementary integration of Structure–Based Drug Design (SBDD) and Ligand–Based drug Design (LBDD) approaches.
Methods based on computation or in silico are currently burgeoning and knowledge-driven, systematically evaluating existing data to explore protein function and develop novel compounds that can modulate its activity. Depending on the availability of protein structures, computational drug discovery techniques are typically SBDD and ligand-based drug design (LBDD). To enhance the success rate of current drug development initiatives, it has been standard practice in the pharmaceutical industry to integrate these approaches in a complementary manner with one another (
Figure 2). When using SBDD, it is necessary to have a three-dimensional (3D) model of the target protein to evaluate and exploit the druggable pockets for screening and creating appropriate ligands, which can subsequently be experimentally confirmed and enhanced. Instead of relying on protein structural data, LBDD uses the information obtained from a wide array of ligands with proven activity to develop prediction models for hit and lead optimization [
126].
Different SB and LB tactics, or a mix of them, might be used at different phases of TB drug design, discovery, and development to mitigate the difficulties associated with experimental techniques. With the availability of the TB genome and proteome and a wealth of structural information, researchers can use big data and molecular simulation to identify potential targets for treatment vs. allows choosing the most promising prospective candidates from a database comprising millions of compounds for a specific TB target. From the validated candidates, a quantitative structural activity relation (QSAR) study is obtainable to understand the mechanism of action and ADMET properties. QSAR facilitates compound development with improved efficacy, as well as pharmacokinetic and pharmacodynamics properties.
The information gathered from this research (both positive and negative outcomes) may be saved and used for additional iteration and technique optimization in designing novel TB drugs in the future. Structure-based vs. produced many anti-tuberculosis compounds with appreciable enzymatic inhibition (Table 2 and Table 3). This study provides an overview of the SBDD process and the current approaches for TB drug development in the modern era. Furthermore, we provide an insight on the machine learning (ML) techniques designed to accelerate the process, procedures, management, and application of large amounts of data in TB drug design.
Table 2. Successful SBVS approaches on anti-Mtb and activities of the best compounds *. A summary of Mtb pathways is available in the supporting information.
| System |
PDB Structures |
Function |
Anti-Mtb Activity |
Ref. |
| L-alanine dehydrogenase |
2VHW |
Biosynthesis of l-alanine |
IC50/35.5 μM b |
[127] |
| L-alanine dehydrogenase |
4LMP |
Biosynthesis of l-alanine |
MIC/1.53 μM |
[128] |
| L-alanine dehydrogenase |
2VOJ |
Biosynthesis of l-alanine |
MIC/11.81 µM |
[129] |
| 7,8-diaminopelargonic acid synthase |
3TFU |
Biotin biosynthesis pathway |
MIC/25 μM |
[129] |
| 7,8-diaminopelargonic acid synthase |
3TFU |
Biotin biosynthesis pathway |
MIC/7.86 μM |
[130] |
| Cyclopropane mycolic acid synthase 1 |
1KPH |
Cell wall |
MIC50/5.1 μM |
[131] |
| l,d-transpeptidase 2 |
3TUR |
Cell wall |
MIC94/25.0 μM
MIC89/0.2 μM |
[132] |
| GlmU protein [58] |
3ST8 a |
Cell wall |
IC50/9.0 μM b |
|
| NAD⁺-dependent DNA ligase A |
1ZAU/1TAE |
DNA metabolism |
MIC50/15 µM |
[133] |
| Flavin-dependent thymidylate synthase |
2AF6 a |
DNA metabolism |
MIC90/125 μM |
[134] |
| Flavin-dependent thymidylate synthase |
2AF6 |
DNA metabolism |
IC29/100 μM b |
[135] |
| DNA gyrase |
4BAE |
DNA topology |
MIC/7.8 µM |
[136] |
| Dihydrofolate reductase |
Mtb: 1DF7; human: 1OHJ |
Folate pathway |
MIC/25 μM |
[137] |
| Salicylate synthase |
3VEH |
Iron acquisition |
MIC99/156 μM |
[138] |
| Transcription factor IdeR |
1U8R |
Iron acquisition control |
MIC90/17.5 μg/ml |
[139] |
| Flavin-dependent oxidoreductase MelF |
2WGK |
Needed to withstand ROS-and RNS-induced stress |
MIC/13.5 μM |
[140] |
| Leucyl-tRNA synthetase |
2V0C |
Protein synthesis |
MIC/25 µM |
[141,142] |
| 3-dehydroquinate dehydratase |
2Y71 |
Shikimate pathway |
MIC/6.25 µg/mL |
[143] |
| 3-dehydroquinate dehydratase |
15 PDB structures |
Shikimate pathway |
MIC/100 mg/ml |
[144] |
| Haloalkane dehalogenase |
2QVB |
Unknown |
Kd/3.37 µM b |
[145] |
Table 3. Structure of identified molecules with the best anti-Mtb activity or enzymatic inhibition.
| Structure |
IUPAC Name |
Enzymatic Inhibition |
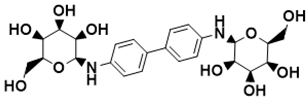 |
(2S,2′S,3S,3′S,4R,4′R,5R,5′R,6S,6′S)-6,6′-([1,1′-biphenyl]-4,4′-diylbis(azanediyl))bis(2-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol) |
Biosynthesis of l-alanine [127] |
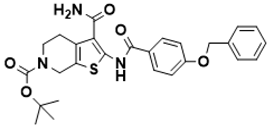 |
tert-butyl 2-(4-(benzyloxy)benzamido)-3-carbamoyl-4,7-dihydrothieno [2,3-c]pyridine-6(5H)-carboxylate |
Biosynthesis of l-alanine [128] |
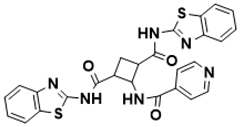 |
N1, N3-bis(benzo[d]thiazol-2-yl)-2-(isonicotinamido)cyclobutane-1,3-dicarboxamide |
Biosynthesis of l-alanine [129] |
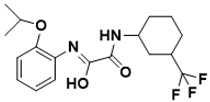 |
(Z)-N-(2-isopropoxyphenyl)-2-oxo-2-((3-(trifluoromethyl)cyclohexyl)amino)acetimidic acid |
Biotin biosynthesis pathway [129] |
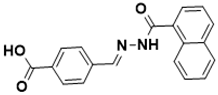 |
(E)-4-((2-(1-naphthoyl)hydrazono)methyl) benzoic acid |
Biotin biosynthesis pathway [130] |
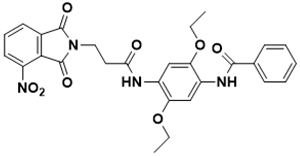 |
N-(2,5-diethoxy-4-(3-(4-nitro-1,3-dioxoisoindolin-2-yl)propanamido)phenyl) benzamide |
Cell wall [131] |
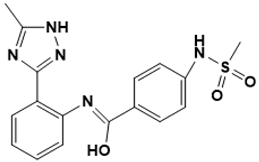 |
(Z)-N-(2-(5-methyl-1H-1,2,4-triazol-3-yl) phenyl)-4-(methylsulfonamido)benzimidic acid |
Cell wall [132] |
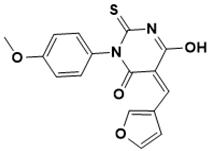 |
(Z)-5-(furan-3-ylmethylene)-6-hydroxy-3-(4-methoxyphenyl)-2-thioxo-2,5-dihydropyrimidin-4(3H)-one |
Cell wall [133] |
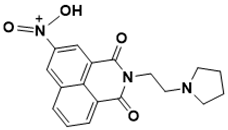 |
N-(1,3-dioxo-2-(2-(pyrrolidin-1-yl)ethyl)-2,3-dihydro-1H-benzo[de]isoquinolin-5-yl)-N-oxohydroxylammonium |
DNA metabolism [134] |
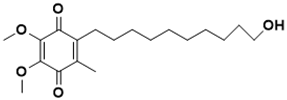 |
2-(10-hydroxydecyl)-5,6-dimethoxy-3-methylcyclohexa-2,5-diene-1,4-dione |
DNA metabolism [135] |
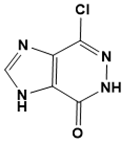 |
7-chloro-3,5-dihydro-4H-imidazo [4, 5-d]pyridazin-4-one |
DNA metabolism [136] |
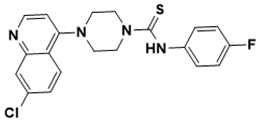 |
4-(7-chloroquinolin-4-yl)-N-(4-fluorophenyl)piperazine-1-carbothioamide |
DNA topology [137] |
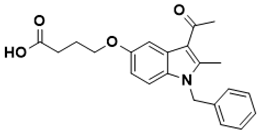 |
4-((3-acetyl-1-benzyl-2-methyl-1H-indol-5-yl)oxy)butanoic acid |
Folate pathway [138] |
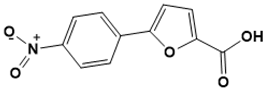 |
5-(4-nitrophenyl)furan-2-carboxylic acid |
Iron acquisition [139] |
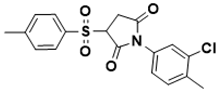 |
1-(3-chloro-4-methylphenyl)-3-tosylpyrrolidine-2,5-dione |
Iron acquisition control [140] |
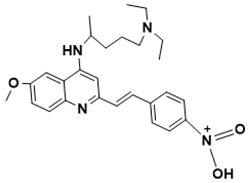 |
(E)-N-(4-(2-(4-((5-(diethylamino)pentan-2-yl)amino)-6-methoxyquinolin-2-yl)vinyl)phenyl)-N-oxohydroxylammonium |
Needed to withstand ROS- and RNS-induced stress [141] |
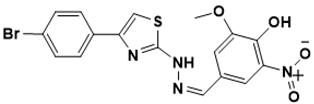 |
(Z)-4-((2-(4-(4-bromophenyl)thiazol-2-yl)hydrazono)methyl)-2-methoxy-6-nitrophenol |
Protein synthesis [142] |
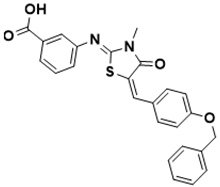 |
3-(((Z)-5-((E)-4-(benzyloxy)benzylidene)-3-methyl-4-oxothiazolidin-2-ylidene)amino)benzoic acid |
Shikimate pathway [143] |
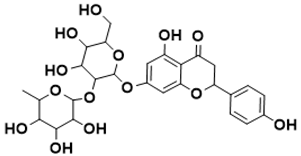 |
7-((4,5-dihydroxy-6-(hydroxymethyl)-3-((3,4,5-trihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)tetrahydro-2H-pyran-2-yl)oxy)-5-hydroxy-2-(4-hydroxyphenyl)chroman-4-one |
Shikimate pathway [144] |
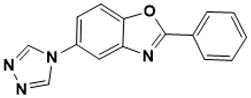 |
2-phenyl-5-(4H-1,2,4-triazol-4-yl)benzo[d]oxazole |
Unknown [145] |
3. Data Application and Management in Tuberculosis Drug Development
Massive data and complex data analysis are the hallmark of the fourth industrial revolution (4IR), profoundly impacting our daily lives’ coordination and conduct. The rise of a big data approach transformed our strategies to deal with age-old challenges in tuberculosis drug development through innovation in cloud data storage and management and improvement in bioinformatics and cheminformatics algorithms. Furthermore, the affordable sequencing technology enables studying all aspects of molecular characters of diseases. Examples are epigenetics, RNA sequencing, metagenomics, targeted sequencing, whole-genome sequencing, and variant detection sequencing [
146]. SBDD and other forms of drug development leverage the analysis of vast biological and chemical data generated and stored on publicly available database repositories in cyberspace [
147].
Considering that tuberculosis is a long-standing disease, volumes of accumulated information await usage to curb this infection. Information on TB drug development is available on the TB Database (
http://tbdb.bu.edu/tbdb_sysbio/MultiHome.html; accessed on 5 September 2021) [
148,
149]. Similarly, Mycobrowser (
https://mycobrowser.epfl.ch/; accessed on 5 September 2021) [
150] contains information on mycobacterium multi-omics. This repository [
150] stores experimental and computational models of TB molecular mechanism pathways and several pathogenic mycobacteria. Mycobrowser also connects with UniProt (
https://www.uniprot.org/; accessed on 5 September 2021) [
151], the most widely used protein database containing mycobacterium protein information. Clinical data on TB are also accessible on the TB Portals (
https://tbportals.niaid.nih.gov/; accessed on 5 September 2021) [
152]. Innovations in structural biology and bioinformatics resulted in an influx of structural data. These interventions led to thousands of 3D protein structures generated from X-ray crystallography, nuclear magnetic resonance (NMR), cryo-electron microscopy (cryo-EM), and homology modeling experiments. PDB [
19], PDBsum [
153], and other structural databases store these research results. Hence, the availability of chemical libraries (
Table 4) was made possible by expanding the digital chemical space [
124] and advancements in chemical synthesis [
154].
Table 4. Accessible public and commercial repositories on TB drug development.
3.1. SBDD Based on Mtb Proteins
The availability of therapeutically important protein 3D structures made SBDD the most desirable approach for drug design and development ahead of ligand-based drug design (LBDD). However, to enhance the success rate of recent drug development initiatives, it has become customary to integrate SBDD with LBDD approaches in a complementary manner. Using the 3D structures of targets to study and exploit the catalytic pocket, SBDD can search and create appropriate ligands that can subsequently be verified and optimized experimentally. To mitigate the difficulties associated with experimental techniques, several types of SB and LB tactics, or a mix of them, might be used at various phases of TB drug design and development. With the availability of TB multi-omics and a large amount of structural biodata, we can use cheminformatics data mining, data engineering, docking, and homology modeling to identify potential targets.
Virtual screening facilitates choosing the most promising prospective ligand(s) from a database comprising millions of identified molecules for a specific tuberculosis target. The output of candidate compound validation using structure-activity (SA) studies enables a better understanding of the mechanism of action and ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties, thus allowing better development of compounds with improved activity and better pharmacological profiles. The information gathered from this research (both good and negative outcomes) may be saved and used for additional iteration and technique optimization in the future design of new tuberculosis drugs. More than 800 CADD software and webservers (free or commercial) are available, hosted by the Swiss Institute of Bioinformatics at
www.click2drug.org (accessed on 5 September 2021). These provide unlimited opportunities to explore drug discovery and design [
1].
Table 5 summarizes some available CADD software.
Table 5. Accessible websites to retrieve software for CADD.
SBDD takes advantage of target protein 3D structure availability. However, if the 3D model of the therapeutically important receptor is not available, computational approaches through homology modeling enable the 3D model prediction of the receptor. Homology modeling or comparative modeling is the most reliable method for 3D protein structure prediction. The methodology entails predicting the 3D structure of the receptor from a homologous protein with at least a 40% similarity index. Threading and ab initio modeling are also methods of protein structure prediction [
15]. After obtaining the 3D structure of the target, it is crucial to validate the model by examining the molecular characteristics in a Ramachandran plot. This metric shows the distribution of the ϕ and ψ dihedral angle conformations of the constituting residues in the receptor structure [
179]. There are several techniques to validate the predicted protein model [
15,
175,
180].
After determining the target structure, the next step is to determine the catalytic pocket. Catalytic or binding pockets are tiny spaces where ligands attach to the target, inducing the intended result. Consequently, it is crucial to identify the most suitable location on the target protein for ligand binding. Even though protein is dynamic in nature, only a few techniques can identify possible binding residues in the binding pocket. Identification of binding sites on a particular target requires the knowledge of interaction energy and van der Waals (vdW) forces. There are many strategies for catalytic site mappings using interaction energy computation through SBDD. This technique can identify locations on the target receptor that interact positively with functional moieties on the drug-like compounds. These approaches find probes that have energetically advantageous interactions with proteins. Q-SiteFinder [
176] is an energy-based technique for predicting catalytic sites widely utilized in the pharmaceutical industry. It is possible to compute the vdW interaction energies of proteins with a methyl probe by using this approach. Those with favored energy values are maintained and grouped in the final product. The total interaction energies of these probe clusters serve as the determinant of their ranking. Aside from that, the functional annotation of interacting protein residues in the binding site allows for the determination of the binding site.
It is also essential to remember that additional possible binding sites, referred to as allosteric sites, may also be present on the target protein surface. Drug development attempts in the conventional sense frequently target the important (orthosteric) binding site to prevent natural substrate binding. Besides, researchers have unveiled noncatalytic sites of Mtb proteins. Shi and colleagues identified a second druggable binding site (allosteric) in Mtb UDP-galactopyranose mutase (UGM) [
181]. MS-208, a well-known Mtb-UGM inhibitor, was categorized as a noncompetitive/mixed inhibitor based on NMR and kinetics investigations. This observation implies that MS-208 binds to another location in the receptor and affects the natural enzyme substrate from recognizing the primary pocket. They [
181] predicted the allosteric sites for MS-208 on the enzyme via docking with AutoDock Vina [
182]. The two identified regions, designated A-site and S-site, show favorable and stable interaction with the ligand after molecular dynamics using Amber [
183]. Simulations facilitate structural and functional relationship determination. Because the A-site-bound structure demonstrated the most stable complex formation with good interaction energy and a higher number of contacts, they hypothesized that this site represents an allosteric druggable binding site in Mtb-UGM [
181].
After appropriate identification of all druggable sites on the receptor, next comes hit discovery, accomplished by docking chemical libraries into the active cavity of the target receptor. Earlier, the routine in lead discovery required choosing a specific collection of ligands that can play a critical role in identifying and optimizing leads [
177]. SBDD blends two distinct approaches for hit search (VS and de novo design) into a single framework.
3.2. Virtual Screening as a Method of Lead Identification
Currently, vs. has emerged as a dynamic and profitable technique in the pharmaceutical business, particularly for prospecting new drug-like compounds or so-called lead identification [
184]. There are two forms of VS: ligand-based vs. (LBVS) and structure-based vs. (SBVS). Biological data is processed in LBVS to distinguish inactive molecules from active ones. Based on consensus pharmacophores, this information facilitates highly functional scaffold identification [
185], similarity, or various descriptors. LBVS produces results that are closely related to known active pharmaceutical ingredients. The procedure involves scanning chemical libraries of structures to find molecules with known like potency or that share a pharmacophore or moiety with known activity. The results are typically positive (pharmacophore substructure similarity search) [
186]. A moiety substructure search requires using the 2D- or 3D-structure of various ligands to find closely related structures. Usually, comparable substances have similar effects when using ligand-based techniques; thus, they are called similarity methods. For example, if one or more active compounds are known, it is feasible to search a database for comparable but more potent compounds [
187].
SBVS allows docking numerous chemical compounds against an enzyme-binding or catalytic site in a short time [
188,
189,
190,
191]. The computer algorithms facilitate target protein docking with one of the vast libraries of drug-like chemicals that are commercially or publicly accessible (
Table 4). Subsequent steps for search refinement are molecular docking, MD simulations, and experimental tests to obtain IC
50 or other efficacy parameters [
192]. SBVS relies on the scoring of ligands to function correctly. In contrast to ligand-based techniques, structure-based techniques do not rely on previously collected experimental data to be effective.
3.3. De Novo Drug Design—A Signature to the Drug Discovery Process
De novo drug design involves creating unique chemical compounds from the ground up, starting with molecular building blocks. The essence of this technique is to design chemical structures of tiny molecules that bind to the target active site with high affinity [
193], then test these structures experimentally. A variation in approach is typically employed when designing from scratch, and the design algorithm must integrate the search space information acquired. Usually, researchers incorporate positive and negative designs with one another. When using the former strategy, a search is to constrain certain regions of chemical space, which increases the likelihood of discovering results with notable characteristics. The search parameters are set in the negative mode to avoid choosing false positives [
194]. Despite its sufficiency in functional scoring analysis, chemical compound design using computational approaches connects organic synthesis but cannot replace it [
195]. It is fundamental in the design stage to conduct a thorough evaluation of candidates’ compounds. One of these evaluation tools is the scoring function; multiple scoring functions for multi-objective drug discovery hybrids [
196] create many different characteristics simultaneously.
De novo drug design is in two categories: (A) ligand-based drug design and (B) receptor/enzyme-based drug design. The latter method is popular currently. Creating appropriate small molecules for enzyme-based design requires high-quality target protein structures and precise knowledge of proteins’ active sites. The approach entails small molecules designed by matching fragment moiety into the target proteins’ binding pockets. The process requires using computer programs or co-crystallization of the ligand with the receptor [
197]. Two ways to execute the ligand-based design are by linking together crucial components, such as atoms or fragments (single rings, amines, and hydrocarbons) to produce an entirely new chemical molecule, or by simply generating ligands from a single parent unit. The fragment-linking technique uses information of the active site to map the likely interaction locations for the different functional groups contained in the design drug fragments [
198,
199]. One must link these functional groups’ moieties to one another to form an absolute compound. The fragment-growing method features fragment development within the active site, monitored by appropriate search algorithms [
199].
These search algorithms make use of scoring systems to determine the likelihood of growth. Fragment-based de novo design is a method of creating new molecules that use the whole chemical space. When using the linking technique, the selection of linkers is imperative. The outside-in strategy and the inside-out approach are both methods for anchoring fragments in the binding site. The outside-in system is the more common method. The outside-in methodology involves the construction blocks placed near or on the edge of the binding site, and the active site gradually expands inward. The inside-out method uses construction pieces randomly placed within the active site region, then constructed outward [
100].
3.4. Molecular Docking and Density Functional Theory Applied to Mtb
Molecular docking has been a prime computational technique of SBVS against Mtb enzymes. The molecular-docking technique was the subject of many published research articles as a tool in drug design against Mtb (
Table 6). According to the Himar1 transposon mutagenesis study conducted by DeJesus in 2017 [
200], the majority of the enzymes targeted by this method are enzymes encoded by crucial genes, with the exclusion of antigens BioA, NarL, 85c, EthR, and LipU. Although this technique assigns nonessentiality to genes based on in vitro growth, it cannot be relied on to determine whether genes are nonessential in vivo [
201,
202]. For instance, the NarL enzyme is necessary for anaerobic survival throughout infection, while BioA is crucial for biotin synthesis during the latency phase of Mycobacterium TB infection [
203,
204]. Furthermore, the EthR protein functions in developing ethionamide resistance and, consequently, survives potentials after drug treatment [
205,
206].
Table 6. Studies involving SBVS molecular-docking approaches against Mtb enzymes.
| Program |
Library of Compounds Screened |
Enzyme (Function) |
Ref. |
| AutoDock Vina |
FDA-approved: DrugBank (1932); eLEA3D (1852) |
MurB and MurE (peptidoglycan biosynthesis) |
[207] |
| |
ChemDiv dataset (135,755) |
DprE1 (arabinogalactan biosynthesis) |
[208] |
| |
NCI; Enamine; Asinex; ChemBridge; Vitas-M Lab (total: 5.6 million) |
InhA (mycolic acid biosynthesis) |
[209] |
| AutoDock 4.0 |
Super Natural II database (570) |
RmlD (carbohydrate biosynthesis) |
[210] |
| CDOCKER |
Enamine REAL database (4.5 million) |
BioA (biotin biosynthesis) |
[203] |
| Frigate |
ZINC database (2 million) |
Antigen 85c (lipid metabolism) |
[211] |
| Glide |
FDA-approved (6282) |
LipU (lipid hydrolysis) |
[212] |
| |
ChEMBL antimycobacterial (30,789) |
DprE1 (arabinogalactan biosynthesis) |
[213] |
| |
FDA-approved (3176) |
PknA (protein kinase) |
[214] |
| |
Preselected from Maybridge database (1026) |
InhA (mycolic acid biosynthesis) |
[215] |
| |
Preselected from DrugBank database (1082) |
AroB (shikimate pathway) |
[216] |
| GOLD |
Drugs Now subset of ZINC database (409, 201) |
EthR (transcriptional regulator) |
[205] |
| GOLD and Plants |
Preselected from Enamine database (2050) |
MbtI (mycobactin synthesis) |
[138] |
| GOLD and RFScore |
Selection from 9 million compounds (4379) |
AroQ (Shikimate pathway) |
[217] |
| UCSF Chimera |
CDD-823953; GSK-735826A |
PyrG and PanK (siosynthesis of DNA and RNA) |
[192] |
Many studies of the different targeted enzymes indicate that many are engaged in either intermediate metabolism or lipid metabolism in Mtb. In addition, DNA and RNA regulatory enzymes and cell wall regulator proteins make up the remaining target proteins. Researchers show at least three SBVS efforts against DprE1 and InhA, with InhA being the most frequently targeted. Studies show that InhA is the ultimate target of both isoniazid and ethionamide once activated [
218]. As a result, InhA provides a validated target whose suppression has an in vivo influence on the survival of Mtb. Also, numerous antimycobacterial medicines target DprE1 in the current anti-TB research pipeline [
29,
59]. PyrG, a newly confirmed TB target, also attracted the attention of researchers [
219]. The chemicals used in most research works (
Table 6) are from generic chemical databases containing millions of compounds, while TB-specific databases and natural product, therapeutic repurposing-focused, and other libraries comprise other chemical compounds reported. As a result, the focus of these early drug discovery initiatives continues to be on totally new drug-like chemical discoveries. The apparent lack of further experimental evidence (in vitro or in vivo) showing compound bioactivity in many of these investigations (
Table 6) is an evident issue that precludes these anticipated compounds from being carried onward [
206].
Another important computational tool in drug discovery is the density functional theory (DFT), which applies to TB research for the investigations of catalytic processes [
220,
221], structure-activity relationship analysis [
222], and inhibitor potency [
192,
223]. Chi and colleagues [
223] adopted DFT in an anti-tubercular study to confirm their first observations of a change in an inhibitor-binding mechanism in the MbtI protein after adding a substituted enolpyruvyl moiety to the parent chemical structure previously generated from isochorismate. From their [
223] observation, there were two distinct binding mechanisms (states 1 and 2) noted in the X-ray crystal structures of MbtI complexed with its inhibitors, indicating that the active site is flexible enough to permit ligand binding. With the aid of Gaussian 09 software application [
224] and a theoretical-level hybrid B3LYP [
225,
226], they computed the global minimum configuration of the (E)-3-(1-carboxyprop-1-enyloxy)-2-hydroxybenzoic acid (AMT), Z-methyl-AMT, and E-methyl-AMT inhibitors complexed in solution. The results revealed that the global minimum geometry of both free Z- and E-methyl-AMT is comparable to its bound geometry (state 2), showing that its arrangement enables binding to MbtI. The computation of conformational entropy quantities for the three molecules indicated that Z-methyl-AMT is the least disorganized. Z-methyl-AMT has a conformational lock provided by the methyl moiety in its structure. Even though a pure Z-isomer has not yet surfaced to distinguish it from the E-isomer empirically, this discovery [
224] justifies the powerful interaction of methyl-AMT to MbtI. It provides further knowledge for the future creation of new and effective MbtI drug-like compounds with the aid of DFT.
Despite the widespread success and popularity of DFT, it has flaws stemming from the approximations employed in its operational mode. DFT is difficult to use for system descriptions mainly composed of dispersion (van der Waals) forces, such as gaseous systems, or systems in which dispersion contributes significantly, such as biomolecular systems [
227]. Thus, numerous research studies examined the incorporation of van der Waals [
228,
229,
230] to improve performance and enhance this technique. In addition to these constraints, the description of global potential energy surfaces of charge exchange excitations [
230] is a prime restriction of DFT use in computational drug design. DFT usage overly favors sophisticated users and requires thorough reviews to determine the level of theory/methods to use for a particular system.