The 3D structure and surface characteristics of proteins and peptides are crucial for interactions with receptors or ligands and can be modified to some extent to modulate their biological roles and pharmacological activities. The introduction of halogen atoms on the side-chains of amino acids is a powerful tool for effecting this type of tuning, influencing both the physico-chemical and structural properties of the modified polypeptides, helping to first dissect and then rationally modify features that affect their mode of action.
- antimicrobial peptides (AMPs)
- structure-activity relationship
- fluoro amino acids
- fluoro-proline
- halogenation
- bromo-tryptophan
- peptides
- α- and β-peptoids
- Introduction
Since 1997, Prof. Victor Hruby has stressed how three-dimensional structure can influence the biological activities of peptides and proteins both in the free state and when engaged in interaction with their receptors/acceptors. If, on one hand, the conformations of the backbone (α-helix, β-sheet, β-turn etc.) are the main determinants for three-dimensional structures, on the other, the arrangement of side chains in three-dimensional χ space (where χ1, χ2 etc. are the torsional angles of the side chain bonds) are determinant for molecular recognition, signal transduction, enzymatic specificity, immunomodulation, and other biological effects (Figure 1 Left). Using computational and experimental methods, it is possible to examine the effects of specific structural modifications in constraining the side chain groups of amino acid residues, or of their mimetics, in χ space [1]. Moreover, to study the physico-chemical features required for the design of novel peptide and peptidomimetic agonists, antagonists, inverse agonists, a special emphasis must be given to the use of conformational (ϕ-ψ space) and topographical (χ space) considerations [2,3] (Figure 1 Right).
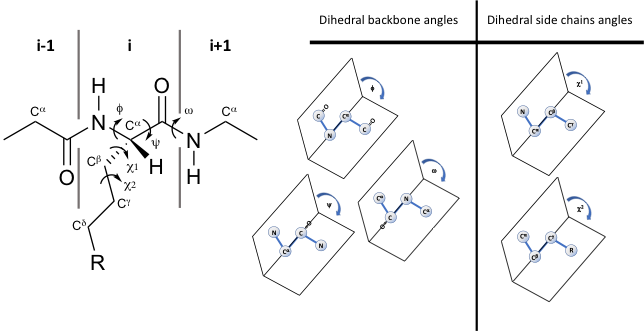
Figure 1. (on left) Representation of the φ, ψ, ω, χ1, and χ2 torsional angles in a peptide, and (on right) their definition as dihedral angles in 3D space, adapted from Ref. [4].
- The Structural and Physico-Chemical Effects of Halogen Atoms in Polypeptides
The introduction of halogen atoms provides many more benefits than other types of substitutions in drugs [9]. In fact, it is estimated that up to one third of drugs undergoing clinical investigation is halogenated [10]. This type of substitution can be used to control the degradability of pharmaceuticals as well as their lipophilicity, membrane permeabilization activity and catabolic stability [11,12]. The ligand-receptor interaction can be modulated by the presence of halogen atoms because of their electron-withdrawing features and their steric-effects [10,13]. This can interestingly find applications from very small molecules to large complexes such as antibodies. For example, halogenated Tyr improved 10 fold the binding affinity of the Fab fragment of an anti-EGF-receptor (059-152) for its antigen [14].
In this role, the halogen atom can also act as an electron donor by allowing the formation of stabilizing interactions such as hydrogen bonds [15,16]. Although it is hydrophobic in nature, the halogen introduces some interesting physico-chemical features not normally associated with this property, such as allowing for the possibility of F···H–N hydrogen bond formation. This is confirmed by through-space coupling in NMR studies [17,18].
The introduction of fluorine into a peptide or a protein offers rich opportunities both when the fluorination site is on an amino acid side chain or on the backbone itself, because of its capacity to locally alter the electronic character of the peptide/protein [19]. The fluorination also has an effect on the folding properties, changing the secondary structure propensity of peptides and protein segments containing aliphatic halogenated amino acids. In addition, fluorine has an impact on the proteolytic stability of peptides, in a manner that depends both on the nature of the fluorinated side chain but also on its effect on the immediate environment [20]. Resistance towards proteolytic digestion due to the introduction of halogenated amino acids cannot however be generalized. It depends on the substitution position and number of introduced halogen atoms on the amino acid side chain. For example, a minimal presence of fluorinated, small side chain amino acids can improve remarkably proteolytic stability [12]. Futhermore, bulky fluorinated residues, such as hexafluoroleucine or trifluoroisoleucine protect polypeptides from proteolytic enzymes. On the other side, fluorinated aromatic amino acids are comparable to their hydrocarbon counterparts in maintaining the proteolytic stability [12]. These observations are rendered even more interesting by studies showing that the introduction of fluorine is relatively non-invasive with respect to structural stability. For example, Kovermann and co-workers investigated the thermodynamic stability and structural integrity of Cold shock protein B from Bacillus subtilis (BsCspB), containing fluorine-labelled phenylalanine or tryptophan residues. Time-resolved fluorescence kinetics was used to monitor chemical denaturation of fluorine-labelled BsCspB, while thermal denaturation was followed by high-resolution 1H and 19F NMR, X-ray [21]. These experiments showed that the presence of F-Phe or F-Trp residue caused only a barely detectable change in thermodynamic stability in comparison to the wild type protein. Moreover, the X-ray structures of fluorinated CBsCspB variants were almost completely superposable with the structure of the wild type protein.
However, since the alignment of C-F bonds with adjacent C=O or C-N bonds is predictable, fluorinated amino acids might be used as a tool to study and control the secondary structure of polypeptides [22]. In this context, 4-fluoroprolines are excellent building blocks to study the pharmacological and structural impact caused by their substitution, especially for the stability and kinetics of protein folding. In this context, the rational substitution of Pro or Hpy with a 4-fluorinated Pro analogue in peptides and proteins can enhance both the thermodynamic and hydrolytic stability, as well as the resistance to unfolding in organic solvents of the engineered protein [23,24].
The pyrrolidine ring of proline introduces a conformational rigidity and constrains into polypeptide backbone, as two of the main chain atoms are constricted on the ring [25,26,27,28]. The pyrrolidine ring adopts predominantly two different conformations. In the endo conformation (Cγ-endo), carbon 4 and α-carbonyl carbon are on the same side of the plane, while in the exo conformation (Cγ-exo), carbon 4 on the pyrrolidine ring points towards the opposite side of the carbonyl group of Pro (Figure 2). The presence of fluorine on the Cγ atom, with its electron-withdrawing properties, can bias the proline ring pucker preferences and “pre-organize” the protein main chain, thus conferring additional stabilization to the whole protein [29,30]. Thus, (2S, 4S)-4-fluoroproline ((4S)-FPro), prefers the Cγ-endo ring pucker and stabilizes the cis peptide-bond conformation, while (2S, 4R)-4-fluoroproline, ((4R)-FPro) stabilizes Cγ-exo ring pucker favoring the trans conformation [28,31,32] (Figure 2).
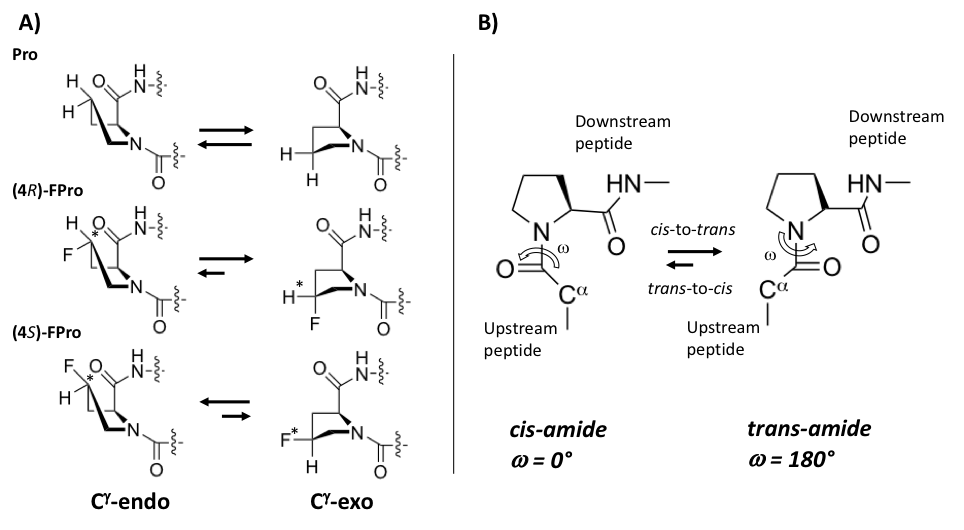
Figure 2. Fluorine effect on proline conformation. (A) pyrrolidine ring puckers in proline, (4R)-FPro, and (4S)-FPro. Fluorine on the Cγ atom introduces a chiral center (indicated by *) with effect on the preference of endo/exo-pucker. (B) cis/trans isomerization by Pro. Figure adapted from Ref. [32].
In folded polypeptides, Xaa-Pro (Xaa = any amino acid) peptide bonds still display preferentially the trans conformation, although in this case both conformations are almost isoenergetic [33]. The cis/trans isomerization of Xaa-Pro bonds is critical during the folding process in polypeptides. The refolding of globular proteins presents an intrinsically slow isomerization step when non-native trans prolyl-peptide bonds in the unfolded state undergo isomerization to reach their native cis state [25]. Therefore, the cis/trans isomerization reaction is typically the rate-limiting step in the native protein folding or refolding processes. The substitution of 10 prolines (9 trans and 1 cis) in the Enhanced Green Fluorescent Protein (EGFP) with (4S)-FPro promoted a faster refolding [25]. However, these results were attributed to several synergistic effects, arising mostly from the adoption of the endo pucker in 9 out of 10 Pro residues, rather than from cis/trans isomerization of prolines.
In 2011, Rubini and co-workers showed that the replacement of Pro 19, Pro 37, and Pro 38 (all displaying an exo pucker and all involved in trans peptide bonds) in human ubiquitin with (4R)-FPro led to the generation of a protein that was 4.71 kJ·mol−1 more stable than the parent protein. Moreover, the introduction of (4R)-FPro did not affect the folding mechanism or biological activity [26]. Recently, Rubini and co-workers studied the impact of 4- and 4,4-difluorinated proline analogues on thioredoxin refolding kinetics. The replacement of the conserved cis Pro76 in the E. coli thioredoxin variant Trx1P with (4S)-FPro [27] led to a 9-fold acceleration of the rate-limiting folding step, i.e. cis/trans isomerization of Xaa-Pro bond. This step occurs in the context of a long-lived folding intermediate (Itrans) that displays an intact tertiary structure with a buried non-native trans Pro76 [34]. Interestingly, the Pro to (4R)-FPro [27] and 4,4-difluoroproline (4,4-FPro) [35] substitution at position 76 in Trx1P did not lead to an increase in the refolding rate (Table 1). The high-resolution crystal structures of Trx1P and of its monofluorinated variants could not explain the difference in the refolding kinetics, as the three structures are almost indistinguishable [36].
Table 1. Kinetic refolding parameters of Trx1P, Trx1-(4R)-FPro, Trx1-(4S)-FPro, and Trx1-(4,4)-FPro.
|
|
k of Itrans→Ncis [s−1]a |
Normalised Rate for |
Ref. |
|
Trx1P |
9.3 ± 0.5 × 10−5 |
1 |
[35] |
|
Trx1-(4R)-FPro |
8.7 ± 2.5 × 10−5 |
0.94 |
[27] |
|
Trx1-(4S)-FPro |
8.0 ± 2.5 × 10−4 |
8.6 |
[27] |
|
Trx1-(4,4)-FPro |
7.9 ± 0.7 × 10−5 |
0.85 |
[35] |
A possible explanation for the folding behavior of these fluorinated protein variants could lie in the transition state of the refolding reaction, instead of in the 3D structure of the native state. The incorporation of (4S)- and (4R)-FPro at cis Pro48 led to similar results for pseudo-wild type barstar protein [37].
Torbeev and Hilvert [38] showed that the incorporation of 4,4-FPro at cis Pro32 into human β2-microglobulin eliminated the rate limiting step for the folding of this protein. The authors suggested that the strong electron-withdrawing effect of the two fluorine atoms on the Pro ring dramatically accelerated the trans to cis prolyl bond isomerization. In fact, in comparison to the 4-monofluorinated Pro isomers, 4,4-FPro shows the lowest energy barrier for cis/trans isomerization; however, its application for the acceleration of folding of globular proteins seems to be context-dependent.
The introduction of (4R)-FPro stabilized the conformation of proline-rich peptides in aqueous solution. The proline-rich sequences in proteins that adopt a polyproline type I (PP I) helical conformation are characterized by the cis orientation for the Xaa-Pro bond, while PP II helical segments display prolyl amide trans isomers. The effect of Cγ exo/endoisomerization of the Pro ring pucker influences the cis/trans conformer ratio of the amide Xaa-Pro bond [28,39,40,41] as recently demonstrated by NMR, X-ray and molecular dynamics (MD) studies [42]. In collagen mimetics, the substitution of (4S)-Hyp or (4R)-FPro for the Pro residues in the sequence stabilizes the Cγ exo pucker and, thus, the PP II helix conformation. On the other hand, the substitution with (4S)-FPro at the Yaa-Pro position caused a relative decrease in thermostability, highlighting the impact of the 4-position stereochemistry [28]. The (4R)-FPro enforces the exo ring pucker, favoring the prolyl amide trans isomer [28,40,43]. With respect to the placement of F-substituted residues, Lin et al. found, using short model peptides, that C-terminal stereoelectronic effects might influence the stability of PP II conformation and the PP II/PP I conversion rate more than the N-terminal effects [44]. All these investigations have demonstrated that the use of FPro is a rational tool to probe polypeptide stability, dynamics, and conformational and structural effects, although some complications were observed possibly due to the chemical and structural constraints imposed by these complex molecules.
The study of structure-activity relationships is now supported also by computational approaches. The application of molecular dynamics (MD) simulations on peptides/peptidomimetics has become an important tool. In particular, the successful prediction of the folding and dynamics of several proteins and peptides, using MD simulations, has significant applications in drug design studies [45,46,47,48]. Their performance is linked to the accuracy of the empirical force fields used in the simulations [49]. Among noncovalent interactions, that are recognized to be important, the halogen bond (or X-bond) is an intriguing tool for engineering protein–ligand interactions and for controlling the structures of proteins and nucleic acids. A structure−activity relationship study between halogenated compounds and a subtype of 5-hydroxytryptamine receptor (5-HT7R) is a good example [50]. The development of new computational modeling tools for X-bonds in biological molecules, has been reported based on a set of potential energy functions that describe the anisotropic electrostatic and shape properties of the halogens participating in them [51,52]. Recently, the force field has been generalized by reducing the number of variables to just one for each halogen type and by estimating the electrostatic variable through a standard restricted electrostatic potential calculation of atomic charges. The simplified force field was validated against the AMBER force field, showing that Rappè et al.’s force field [53] is more adaptable for incorporation into classical molecular mechanics/dynamics algorithms, including those commonly used to design inhibitors against therapeutic targets in medicinal chemistry and materials in biomolecular engineering. Halogenation, and in particular fluorination, can improve several features of proteins, such as thermal and proteolytic stability and/or their enzymatic activity. A study of the impact of fluorination on hydrophobicity was recently carried out using dynamics simulations, together with a new fixed-charge, atomistic force field, to quantify the changes in hydration free energy, ΔGHyd, for amino acids with alkyl side chains and with 1 to 6 -CH → -CF side chain substitutions [54]. The results underline two main contributions traceable to alteration of side chain-water interactions and of the number of backbone-water hydrogen bonds. In conclusion, in recent years, many design tools have been developed to mechanically understand how fluorination plays a role in structure-activity relationships. This knowledge can be applied downstream in several ways, ranging from the effect on the hydrophobicity of (bio) polymers, to electrostatic properties.
- Halo-Amino Acids and Halogenated Non-Ribosomal Peptides
Most of the molecular types belonging to the halo-amino acids of natural origin contain halogens bound to sp2 carbons. The Csp2-X bond (X = Br, Cl) is less prone to react with nucleophiles, for example water, but also amino groups, enolates or hydroxyls, which are ubiquitously present in living matter. For this reason, the most representative examples belong to derivatives of the aromatic amino acids: tyrosine, histidine and tryptophan. Aliphatic chlorinated non-proteinogenic amino acids, which are less reactive compared to their corresponding bromides, have also been identified in nature, where both Csp2-Cl and Csp3-Cl bonds were observed. Finally, it should be noted that halo-amino acids have not only been found in peptides, but also provided a key starting material for the assembly of complex natural products [55].
3.1. Halogenated Tyrosines and Histidines
Sponges of the order Verongida produce bioactive halogenated amino acids. These compounds were included in the chitin-based skeletons of the demosponge Aplysina cavernicola (Figure 3). Specifically, 3-chlorotyrosine (1 in the Figure 3), 5-bromohistidine (2), 3-bromotyrosine (3), 3,5-dichlorotyrosine (4), 3-iodotyrosine (5), 3-bromo-5-chlorotyrosine (6), 3,5-dibromotyrosine (7), 3-chloro-5-iodotyrosine (8), 3-bromo-5-iodotyrosine (9), 3,5-diiodotyrosine (10) were identified by electron impact (EI)-MS spectrometry in comparison with known samples [56]. 3-Bromo-5-chlorotyrosine (6) was also previously isolated and fully characterized from the gastropod mollusk Buccinum undatum [58].
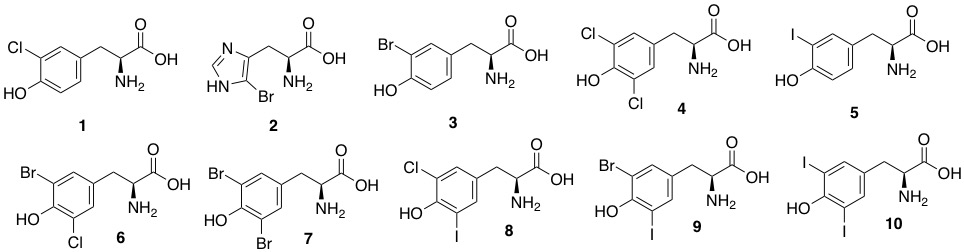
Figure 3. Natural halogenated tyrosines and histidine. In a recent study, tyrosines have been shown to be the preferential site of halogenation of peptides, variously leading to formation of compounds (1) and (3–10).
The compounds (1) and (3)–)10) were found to be variously bioactive as antibacterial, anticancer, antiparasitic, antiplasmodial, antiinflammatory, and antifouling molecules [59,60,61,62,63,64].
3.2. Halogenated Tryptophans
Five regioisomeric bromotryptophans (BrTrps) (15–19 in Figure 4) have been identified in sponges and lower marine invertebrates [67]. Compounds 15–19, are formed by post-translational modifications, i.e., bromination of tryphtophans after they are inserted in complex macromolecules of peptidic, macrocyclic or alkaloid nature. The relative abundance of Br in sea water (0.9 mM) and the presence of broad spectrum bromo- or lactoperoxidases in marine organisms are likely responsible of a plethora of brominated compounds in the marine flora and fauna, including peptides. All of the compounds (15–19) have been found in peptides from marine organisms. However, they have never been isolated as free amino acids, and when tested in this form for bioactivity, this was negligible.
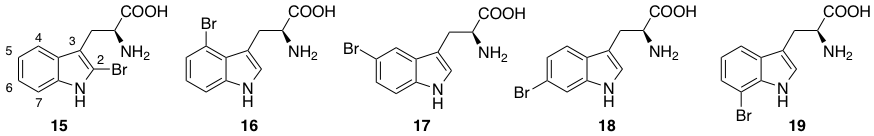
Figure 4. Naturally occurring halogenated tryptophans.
Conversely, peptides containing the halogenated amino acids (15–19) showed significant pharmacological activities. Notably, 2-BrTrp (15) is present in cyclic peptides isolated from marine sponges and endowed with potent bioactivity. The presence of natural peptides with brominated tryptophans have made these residues a topic of considerable interest for in vitro studies. As a consequence, their synthetic production for downstream applications has also been investigated. Bittner et al. provide a comprehensive review of the synthetic methodology required to prepare brominated tryptophan, as shown in Figure 4, (15–19) [67], and bioenzymatic approaches towards halogenated aromatic amino acids have also been recently reported [75,76].
The interest in non-native amino acid building blocks is further fostered by their importance in protein research and engineering [77]. Hence, noncanonical amino acids, endowed with peculiar catalytic activity have been synthesized for introduction into peptides by solid-phase synthesis, or into proteins by mean of recombinant technologies [78,79].
3.3. Aliphatic Halogenated Amino Acids
Aliphatic halogenated amino acids have also been described. γ-chloronorvaline (20, Figure 5) was identified from cultures of the microbe Streptomyces griseosporeus and it was found to possess antibacterial activity, especially against Pseudomonas aeruginosa [82]. The novel 4-amino-3-chloro-2-pentenedioic acid (21) was found in Streptomyces viridogenes [83], and is also produced by Streptomyces xanthocidicus [84,85]. Isoxazoline U-43,795 (22), displaying anticancer activity, was isolated from cultures of Streptomyces sviceus, and its anticancer activity was established [86]. 4-ClLys ((2S,4R)-2,6-diamino-4-chlorohexanoic acid, 29) [5] was recently found to be an intermediate in the production of β-ethynylserine (βes), whose biosynthesis is initiated by chlorination of L-lysine at the Cγ position by the enzyme BesD halogenase. Several chlorinated amino acids are present in fungi [87].
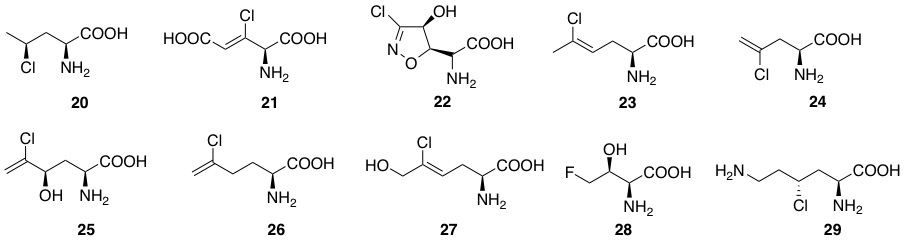
Figure 5. Natural aliphatic halogenated amino acids.
3.4. Halogenated Amino Acids in Non-Ribosomal Peptides
Halogenated amino acids are well represented in several non-ribosomal peptides. These compounds are synthesized, at least in part, by one or more specialized non-ribosomal peptide synthetase enzymes, encoded by genes usually organized in a single operon in bacteria or in a gene cluster in eukaryotes [91].
3.5. Synthesis of Naturally Occurring and Artificial Halogenated Amino Acids
Most halogenated natural products are usually brominated or chlorinated, in a way that depends on the abundance of these elements in the environment in which they are generated. In particular, brominated products in general, including amino acids, are largely found from marine organisms, as reported in the paragraph above [99,100]. On the other hand, the biosynthesis of fluorinated natural products is relatively rare, despite the natural abundance of fluorine. The reason for this could be due to both its high electronegativity and the high enthalpic cost associated with the fluorine desolvation [101]. The introduction of polar C-halogen bonds deeply impacts on the potential bioactivity of natural organic compounds. Fluorination makes no exception and a significant effect has often been reported on introduction of fluorine into bio-organic compounds. The native introduction of fluorine by enzymatic processes is still not well understood, although some enzymatic reactions have been described for some bacteria, such as Streptomyces cattleya, and it is used in chemoenzymatic synthesis [102,103]. There is an ongoing interest in understanding the mechanism by which halogenation occurs. This may help to design or optimize strategies to synthesize halogenated amino acids. For example, several studies have focused on the use of fluoro-pyruvate as a substrate for type II aldolases, to synthesize some chiral organo-fluorines [103]. The demand of halogenated amino acids led to development of several artificial strategies to provide these compounds for further rational design of more complex molecules.
The synthesis of halogenated amino acids has already been reviewed and the excellent chapter by Strickland and Willis remains an important reference [104], as well as the methods of preparation of brominated tryptophan (15–19) reported by Bittner et al. [67].
- Halogenated AMPs
4.1. AMPs as Novel Drug Candidates and Their Mechanism of Action
Antimicrobial peptides (AMPs) are compounds that can be potentially used as al-ternatives to conventional antibiotics [123] or effective adjuvants that can help conven-tional antibiotics to overcome resistance [124–129]. However, AMPs still have critical issues that prevent them from clinical application and require further optimization. Therefore, these compounds represent an intriguing opportunity to observe how halo-genated amino acids may tune some physico-chemical properties of AMPs that are re-sponsible of various aspects of their biological activity.
Endogenous AMPs, often referred to also as host-defense peptides (HDPs) [130], are ancient, gene-encoded, innate immune effectors. They are ubiquitously found in both prokaryotic and eukaryotic organisms and display a remarkable level of structural and functional diversity. In prokaryotes, they play a role in competition for resources with other strains in nutrient-poor environments. In eukaryotes they have a fundamental role in keeping commensal organisms under control and as a first line of defense against microbial infections [131,132].
In addition to a direct antimicrobial activity, many AMPs also exert immunomodulatory or healing-promoting activities, making them interesting molecules for the development of novel therapeutics [133,134]. The mechanism of action of AMPs, unlike most of the conventional antibiotics, is not usually based on a specific molecular target since AMPs interact directly with microbial membranes on which they exert a destabilizing and damaging effect known as “lytic” [147–149]. Their structural properties (amphipathicity, cationic charge, shape, and size) allow them to interact with and efficiently permeabilize [150] the microbial lipid bilayer. Interaction with the target cell membrane can be described by five distinct steps: (1) contact with cell surface of target cells based on physical affinity (electrostatic and/or hydrophobic interactions); (2) conformational rearrangement at the membrane surface and membrane insertion; (3) accumulation up to a threshold level; (4) an alteration of the membrane occurs by either permeabilization or depolarization or causing any other membrane misfunction (detergent effect, micellization, etc.) through a direct or indirect abnormality (this step may involve peptide oligomerization); and (5) killing of the microorganisms through a lytic process of the membranes or any other irreversible processes within the cell [92].
The secondary structure is a fundamental feature of AMPs, with helical or β-sheet stretches adopted or arranged for optimal insertion into the membrane bilayer. As a consequence of this generalized disruption and lack of specific targets, bacteria have difficulty to develop permanent resistance against AMPs. However, the use of this new class of antibiotics is mainly limited by in-vivo toxicity, as they can also interact with (and damage) animal cell membranes to some extent. Other problems are related to the peptidic nature of these molecules, including poor bioavailability due to susceptibility to proteases and rapid clearance, potential antigenicity, and the high cost of production. To address some of these limitations, some strategies involve incorporation of non-proteinogenic amino acids [159,160], which are useful tools for modulating essential AMP-like properties based on native AMPs. They can be used to optimize size; backbone stability and/or conformation; side-chain properties, such as hydrophobicity or charge; and overall properties, such as amphipathicity. Other strategies are based on the design of hybrid peptides [161–163] to discover innovative drug leads with high cell selectivity and/or improved bioavailability [164,165].
4.2. Synthetic Halogenated AMPs
Native AMPs and their synthetic analogues have received increasing attention from academic research and the pharmaceutical industry [182]. However, despite their promising antibiotic properties, they suffer from several disadvantages as peptide therapeutics, for example, the susceptibility to proteolytic degradation, the pH- and/or salinity-dependence of their activity, loss of antimicrobial effect due to binding to serum components [183,184], as well as a potential toxicity to mammalian cells. Moreover, the costs of industrial production are high. For these reasons, modifications to AMPs have been developed with the aim to improve antimicrobial potency and stability while at the same time to reduce toxicity and size. This can be done by chemical modifications, such as substitution of one or more amino acid residues of the natural AMP template with other L- or D-amino acids, both proteinogenic or non-proteinogenic, as well as N-terminal acet-ylation, C-terminal amidation, peptide cyclization (usually via disulfide bond formation and more rarely by backbone cyclization), or introduction of unnatural amino acids to synthesize α-peptoids [185], β-peptoids [186], β-peptides [187], peptide/peptoid hybrids [188–190], and/or functionalized with lipo-amino acids [191] or with PEG [192].
Fluoro-amino acids are introduced into peptides or proteins as internal probes to study inter and intramolecular interactions as well as for structural stability and dynamics measurements [193–195]. The fluoro-amino acids are a useful tool to explore their mechanisms of action, as the Fluorine-19 nucleus constitutes an excellent NMR probe due to a large chemical shift dispersion, high sensitivity to its local environment, and low background noise caused by the absence of fluorine in biological molecules [196,197].
A recent study [203] on tritrpticin and its amidated analog tritrp1, which are AMP members of the Trp/Arg-rich family with a strong antimicrobial activity against bacteria and fungi [204], showed that fluorination preserved the antimicrobial activity as well as the mechanism of action.
With respect to α-peptide/β-peptoid hybrids, in recent studies, the influence of hydrophobicity, fluorination, and distribution of cationic/hydrophobic residues on antimicrobial, hemolytic, and cytotoxic properties were investigated [212,213].
- Future Perspectives and Conclusions
This review has been focused on the structural and biological effects of introducing halogen atoms into amino acid residues. Special emphasis was placed to natural and synthetic peptides with antimicrobial activity. Primarily, we showed that halogenation can affect the structure and in particular the conformations that can be adopted by peptides as well as protein folding. Because of particular physico-chemical properties of halogens, their use in modified amino acids therefore becomes a powerful tool to study the mechanisms and dynamics of these processes. Furthermore, halogenation, such tryptophan bromination, resulted to be useful to optimize [222] the characteristics of bioactive molecules with pharmaceutical activity, as shown for products from marine organisms. Fluorine is efficaciously used as an internal probe for F19-NMR studies of peptide and protein structure-activity relationships (SAR), especially in interaction with biological membranes [18,223]. SAR studies have also revealed a correlation between the enhanced hydrophobicity resulting from halogenation and the antimicrobial activity of AMPs and their peptidomimetics. This could help to design such molecules with a finely tuned balance between the charge and hydrophobic features to optimize the pharmacological profile [224,225]. It has been recently shown that halogenated amino acids are also tools for other applications in pharmaceutical and organic chemistry. Bromotryptophan can be, in fact, utilized as a scaffold for the synthesis of new organic compounds [226]. Different types of halogenated Trp, prepared by employing specific halogenases (e.g., marine haloperoxidases) or haloindoles during biosynthesis by tryptophan synthase, can be introduced into peptides or proteins to tune biocompatibility and activity [227]. Moreover, the use of halogenated Tyr has been re-cently shown to increase the binding affinity of an antibody towards its antigen [14].
In conclusion, this review aims to highlight how biological and chemical methodologies can interact very constructively in the study of bioactive peptides. An example is the recent developments of halogenated peptidomimetics built from natural AMP templates. The present work would like to address both the biologist and the chemist who begin to approach AMPs and inform them of the potential of peptide and protein halogenation in a spirit of multi-disciplinarity that is typical of both the academic and research activities of prof. V. Hruby.
This entry is adapted from the peer-reviewed paper 10.3390/molecules26237401