Glioblastoma (GBM) is the deadliest and most aggressive neuroepithelial cancer of the central nervous system (CNS) with an abysmal median survival of 14.6-month despite the multiple forms of intervention. In the United States, the total annual incidence rate of glioma has been ~6 cases per 100,000 individuals, of which GBM accounts for about 50% of the cases, with a higher predominance in males. Clinical studies have indicated that most of the GBM patients present an intact blood–brain barrier (BBB) for certain brain regions, capable of blocking the delivery of agents to cancer sites. The BBB is considered to prevent diffusion of 98% of small-molecule and 100% of large-molecule agents into the brain from blood circulation. Given the aggressive and heterogeneous nature of GBM and the blocking capability of BBB, a very limited number of medications for patients with GBM is available in clinics. In addition, due to the existence of other cellular and extracellular barriers, as well as the development of drug resistance over the treatment course, the efficacy of many current therapeutic approaches has been compromised.
Currently available standards of care for GBM include maximal tumor resection followed by radiotherapy, chemotherapy, and corticosteroids, all of which have immune suppressive characteristics. Unfortunately, complete surgical removal of the whole tumor is almost impossible due to their diffusive characteristics into normal brain tissue. Some reports indicated that ~65% of the post-surgery cases still showed residual tumor cells, which eventually contributed to a high relapse rate of GBM . Therefore, GBM patients may undergo repeated surgical resection, radiotherapy, chemotherapy, or additional bevacizumab treatment. Eventually, most of the patients suffering from GBM will relapse despite an ample set of interventional approaches. According to the data from Surveillance and Epidemiology, the median overall survival (OS) of GBM patients was normally less than 2 years from the time of first progression or relapse. An international phase III randomized trial, conducted by the European Organization for Research and Treatment of Cancer/National Cancer Institute of Canada (EORTC/NCIC), has shown that the median OS of GBM patients who received radiotherapy and Temozolomide therapy remains poor (14.6 months). Moreover, Grossman and colleagues found that the utilization of systemic chemotherapy and hyperfractionated radiation therapy with corticosteroids is likely to disable immune activity. Immune-suppressive characteristics, high toxicity, and lower OS of traditional care made a considerable number of GBM patients (~50%) not accept any second-line of anti-tumor treatment. In addition, there is no evidence that traditional intervention can significantly impact the OS rate under a recurrence setting. Accordingly, given the poor prognosis and limited therapy regimens for patients affected by GBM, there is an urgent need to develop novel therapeutic approaches.
- glioblastoa
- immune-checkpoint inhibitors
- tumor microenvironment
- tumor-associated macrophages and microglia
- immune-related adverse events
1. Immune Microenvironment of Glioblastoma
It has been regarded that the CNS lacks dedicated lymphatic channels for a long time. The CNS was considered as an immune privileged system, devoid of any immune cells. This overstated historical notion was mainly based on the experimental data reported by Peter Medawar, where foreign grafts transplanted into the brains of rodents did not induce any immune response and the same foreign grafts transplanted into other tissues or organs were rejected [1][2][3][4]. However, this perception has been challenged recently since several studies showed vigorous immunosurveillance and meaningful immune response in the CNS [5][6]. For example, the discovery of a novel route of lymphatic-based channels, reported by Louveau and colleagues [5] in 2015 and the findings of robust immune responses in multiple inflammatory conditions [6] have both demonstrated the CNS as a region for active immunosurveillance. Such findings prompted an increase in studies for the feasibility of cancer immunotherapy towards brain tumors. Although immunotherapy holds great potential for treatment of malignant GBM, unique GBM-associated immune suppression and immune escape still provide challenges to generate efficient anti-tumor responses [4][7]. GBM can form a highly immunosuppressive milieu, mediated by distinct immune or tumor cells (Figure 1). Tumor cells normally express plenty of immunosuppressive factors, such as programmed cell death 1 ligand 1 (PD-L1) and indolamine 2,3-dioxygenase (IDO), while reducing antigen presentation by diminishing major histocompatibility complex (MHC) expression [4]. Notably, gliomas produce IDO, whose function relates to the recruitment of regulatory T (Treg) cells and the inhibition of effector T cells through tryptophan depletion [4][8]. In the context of microglial cells, these often secrete transforming growth factor β (TGFβ) and/or interleukin 10 (IL-10) to decrease the amount of myeloid and lymphoid immune cells to boost systemic immunosuppression [4][9][10] (Figure 1). The lymphoid compartment also mediates immunosuppressive effects with Treg cells through upregulation of different soluble factors and some immune-checkpoint molecules [4]. These immunosuppressive factors may ultimately block T-cell proliferation and activation. One unique factor of GBM is its relatively low tumor mutational burden (TMB) which reduces the responding T cell clones resulting in poor adaptive immunity [11][12][13]. High TMB often suggested as a reliable biomarker for ICIs [11][12][13]. Other variables, including chemotherapy, corticosteroids, and patient age-related factors may also lead to immunosuppression in GBM patients [10]. Overall, GBM is considered as a highly immunosuppressive CNS-related tumor.
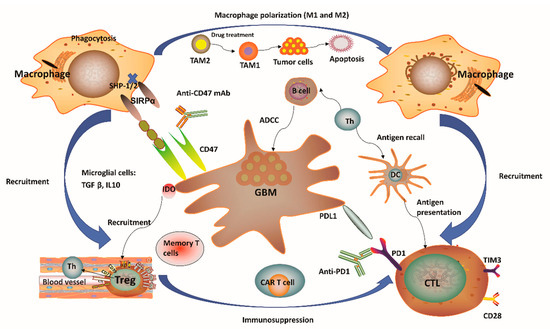
Figure 1. Immunity-related microenvironment of glioblastoma. (1) The immune microenvironment involving glioblastoma (GBM) is characterized by large amounts of CD8+ and CD4+ T cells, M1 and M2 polarized macrophages, microglia, and regulatory T (Treg) cells in addition to a limited number of natural killer (NK) cells. Tumor-associated macrophages and microglia (TAMs) have considerable plasticity toward anti-tumor M1 (inflammatory TAMs) and pro-tumor M2 (anti-inflammatory TAMs) phenotypes. Pharmacological strategies to re-educate tumorigenic M2 TAMs to tumoricidal M1 TAMs may help to relieve immune suppression in the tumor microenvironment (TME), as well as enhance the related anti-tumor activity. (2) GBM normally expressed high levels of immunosuppressive factors, such as programmed cell death 1 ligand 1 (PD-L1) and indolamine 2,3-dioxygenase (IDO), while limiting the presentation of antigens by decreasing major histocompatibility complex (MHC) presentation. The application of IDO inhibitors has effects on Treg cell accumulation. (3) CD47 is highly expressed in various types of tumors. Signal regulatory protein α (SIRPα) is an inhibitory receptor expressed on macrophages and other myeloid immune cells. Upon CD47 binding to SIRPα, src homology 2 domain-containing protein tyrosine phosphatase 1 (SHP-1) and SHP-2 phosphatases are activated to further abrogate phagocytosis via downstream mediators. Disruption of the CD47/SIRPα axis using anti-CD47 antibody (CD47 Ab) can interrupt the inhibitory signaling mediated by SIRPα, thereby promoting phagocytosis of tumor cells. (4) T-cell immunoglobulin and mucin domain-containing protein-3 (TIM3) is a strong negative regulator of lymphocyte function and survival, acting as a marker of CD4+ and CD8+ T-cell exhaustion similarly to programmed cell death 1 (PD-1). It has been verified that the co-expression of PD-1 and TIM3 in lymphocytes is positively correlated with the tumor grade, but it is negatively correlated with progression-free survival (PFS) in different types of tumors including GBM. (5) In the context of microglial cells, these often secrete transforming growth factor β (TGFβ) and/or interleukin 10 (IL-10) to decrease the amount of myeloid and/or lymphoid immune cells, resulting in a systemic immunosuppression and immune evasion of GBM cells. Th, helper T cell; ADCC, antibody-dependent-cell-mediated cytotoxicity; Treg, regulatory T cell; CTL, cytotoxic T lymphocyte; CAR T, chimeric antigen receptor T cell; DC, dendritic cell.
One distinctive aspect of the brain’s microenvironment is related to the bulk of myeloid cell population, which is capable of manipulating the immune microenvironment and GBM progression by producing immunosuppressive and anti-inflammatory cytokines and growth factors, as well as promoting T-cell apoptosis, thus suggesting a new strategy for immunotherapy [14][15]. A considerable population of brain myeloid cells are microglia, which are equivalent to macrophages from other tissues [16]. In the absence of any inflammatory stimulation, the microglia normally arise from the yolk sac and are maintained by continuous replication during our whole life [17]. Upon pro-inflammatory stimulation in the GBM tissue, microglial cells may undergo significant phenotypic changes, while extensive additional macrophages can also be recruited from peripheral monocytes into the tumor site [18][19][20]. Notably, the GBM microenvironment has a surprisingly high composition of tumor-associated macrophages and microglia (TAMs), ranging between 30 and 50% [21] of tumor mass. Such a percentage of TAMs is much higher than the ones observed in other major malignancies such as melanoma [22]. One notable feature of TAMs is that they have considerable plasticity toward anti-tumor M1 (inflammatory TAMs) and pro-tumor M2 (anti-inflammatory TAMs) phenotypes (Figure 1). Redirecting TAMs from immunoinhibitory M2 to immunostimulatory M1 phenotype is a promising approach to elicit an immune response and to inhibit GBM progression since this can reduce immunosuppressive restrains and thus boost immunity driven by cytotoxic T lymphocytes (CTLs) [23]. More recently, research evidence has indicated that pharmacological inhibition provided by certain soluble factors, such as colony-stimulating factor-1 receptor, can dramatically decrease M2 polarization and significantly improve OS [23]. Moreover, several reports have confirmed a strong association between the survival of high-grade glioma patients and M1 or M2 polarization. For instance, M1 polarization has been positively correlated with improved patient survival [24]. In contrast, M2 polarization (assessed by F11R marker) has been negatively correlated with patient survival [25]. Therefore, strategies to target TAMs have emerged as alternate routes for GBM therapy [26]. In this sense, a number of studies have pursued ways to (i) inhibit monocyte recruitment into the CNS, (ii) deplete M2 TAMs, and (iii) reprogram tumorigenic M2 to M1 phenotype [27]. One recent report has also demonstrated TAM-mediated resistance of programmed cell death 1 (PD-1) immunotherapy, thus providing a strong rationale towards TAM targeting as a reliable approach to enhance PD-1-inhibitor treatment response [28]. Of note, TAM-targeted immunotherapy has received particular attention in recent years although investigations related to this promising therapeutic area are still in progress.
2. Overview of Current Immunotherapy Modalities for Glioblastoma
As a paradigm shift in cancer treatment, immunotherapy has recently gained enormous attention and also achieved a rapid expansion in the context of GBM. Immunotherapy approaches for GBM have been focused on ICIs, oncolytic viruses, chimeric T-cell receptors, and dendritic cell (DC) vaccines [4][9]. Figure 2 outlines four distinct immunotherapy modalities available for GBM. We can notice that a successful vaccine for GBM treatment depends on DC-mediated presentation of GBM-related antigens as well as peptides for T-cell activation in the adaptive immune system. Among the pathways involved in these processes, one is related to the combination of T-cell receptors and MHC, while another pathway involves the interaction between CD80/CD86 and CD28. Cytotoxic T lymphocytes (CTLs) can be subsequently activated to kill GBM cells having specific antigens for MHC I presentation [4][29]. In general, tumor cells avoid this disruption by upregulating PD-L1, which binds to its complementary receptor, PD-1 along the T-cell surface to further inhibit the activation of CTLs [30]. We can utilize different approaches of immune-checkpoint blockage to effectively prevent the interaction between PD-L1 and PD-1 in GBM. However, a phase III trial result to compare therapeutic efficacy of nivolumab and bevacizumab in recurrent GBM was disappointing with no improvement in OS (Clinical trial identifier: NCT02017717, Table 1). Cytotoxic T lymphocyte protein 4 (CTLA-4) is another important immune regulatory molecule that binds to CD80 or CD86 and inhibits their combination with CD28 to prevent T-cell activation [30]. Epidermal growth factor receptor variant III (EGFRvIII), IL-13 receptor subunit-α2 (IL-13R α2), and human epidemic growth factor receptor 2 (HER2) are expressed on the surface of GBM cells and may also be targeted by a genetically modified chimeric antigen receptor (CAR) T cell to promote GBM cell death [4][9][29]. Given the promising role of cancer immunotherapies towards GBM pathophysiology, a substantial number of clinical trials have been performed or planned to explore the potential roles and efficacy of targeting these three antigens [4][9][29] (Table 1). These clinical trials have demonstrated feasibility, safety, and efficacy of CAR T cell therapy for GBM. For example, treatment constituted of virus-specific T cells (VSTs) expressing HER2-specific CAR (HER2-CAR VST)in progressive GBM patients resulted in an median OS of up to 11.1 months from the first T cell infusion and 24.5 months from the first diagnosis (Clinical trial identifier: NCT01109095, Table 1).Interestingly, genetic engineering has also been applied in oncolytic viral treatment to produce viruses that may infect tumor cells, trigger tumor cell lysis, and hijack tumor cell replication, which ultimately leads to tumor cell death [4][9] (Table 1). This particular treatment has enabled the breakage of shackles from many tumors and also triggered a higher immune backlash, thus shifting GBM from cold to hot tumor types [4][9]. Promisingly, data from phase II trial have verified the high clinical response in GBM patients after intratumoral inoculation of Polio/Rhinovirus Recombinant (PVSRIPO), with an increase in OS up to 12.5 months from the time of inoculation and higher survival rate at 24 and 36 months over historical controls (Clinical trial identifier: NCT01491893, Table 1). These results show that oncolytic-based therapy has a high potential to improve OS and quality of life for patients affected by GBM [29]. Although oncolytic-based therapies may provide significant immunostimulatory effects, including the depletion of regulatory T cells, the induction of immunogenic cell death, and abscopal effects, these therapeutic approaches still carry some intrinsic limitations. For instance, pro-inflammatory responses caused by oncolytic viruses may potentially limit the application of oncolytic viruses as a single-modality immunotherapy [29]. Besides, CAR T-cell treatment for GBM relies on the identification of stably expressed and sufficient tumor-related antigens, which might eventually limit the clinical application of this therapy [9][29]. Considering the highly heterogeneous characteristics of GBM, one could postulate that targeting one antigen in GBM might not be sufficient to eradicate all the GBM cells. Overall, these obstacles have promoted the development of alternate immunotherapy modalities, which may better recapitulate tumor immunology with improved accuracy.
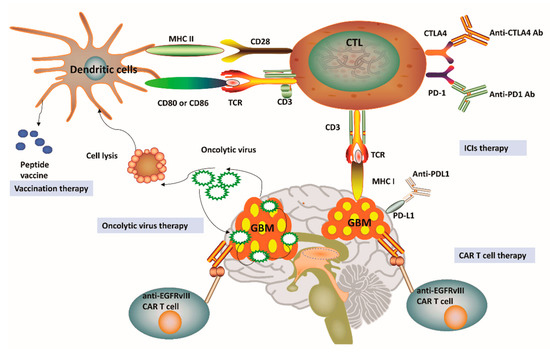
Figure 2. Current immunotherapy strategies for glioblastoma. (1) Vaccines for glioblastoma (GBM) treatment have been relied on dendritic cell (DC)-mediated presentation of GBM-related antigens and peptides for T-cell activation in the adaptive immune system. (2) The immunosuppression status of cytotoxic T lymphocytes (CTLs) can be relieved by the application of immune-checkpoint inhibitors (ICIs), including anti-programmed cell death protein 1 (PD-1), anti-cytotoxic T lymphocyte protein 4 (CTLA-4) and anti-programmed cell death 1 ligand 1 (PD-L1). (3) Genetically engineered chimeric antigen receptor (CAR) T cells can generate artificial T-cell receptors with high affinity to cancer-specific antigens. (4) Genetic engineering has also been applied in oncolytic viral treatment to medicate cancer cell lysis and promote tumor necrosis. MHC, Major histocompatibility complex; TCR, T-cell receptor; EGFRvIII, Epidermal growth factor receptor variant III.
Table 1. Recent clinical studies with immune-checkpoint inhibitors and some combinational therapies targeting glioblastoma.
| Therapeutic Approach | Immune Targets | Type of Glioma | Type of Study | Number of Subjects | Overall Survival (OS) | Progression Free Survival (PFS) | Clinical Trial Identifier | Ref No. |
|---|---|---|---|---|---|---|---|---|
| Immune-Checkpoint Inhibitors and Combinational Therapies | ||||||||
| Nivolumab vs. Bevacizumab | PD-1 | Recurrent glioblastoma | Phase III | 369 | 9.8 vs. 10.0 months | 1.5 vs. 3.5 months | NCT02017717 | [31] |
| Neoadjuvant Nivolumab | PD-1 | Primary and recurrent glioblastoma | Phase II | 30 | 7.3 months | 4.1 months | NCT02550249 | [32] |
| Nivolumab + Radiation + Bevacizumab | PD-1 | Recurrent glioblastoma | Phase II | 94 (recruiting) | N/A | N/A | NCT03743662 | [33] |
| Pembrolizumab | PD-1 | Refractory high grade glioma | Retrospective study | 25 | 4 months | 1.4 months | N/A | [34] |
| Nivolumab + Temozolomide | PD-1 | Glioblastoma | Phase II | 102 (recruiting) | N/A | N/A | NCT04195139 | [35] |
| CAR T Cells ± Nivolumab and Ipilimumab | PD-1, IL13Ralpha2, CTLA4 | Recurrent or refractory glioblastoma | Phase I | 60 (recruiting) | N/A | N/A | NCT04003649 | [36] |
| Ipilimumab + Nivolumab | PD-1 and CTLA4 | Glioblastoma | Phase I | 6 | N/A | N/A | NCT03233152 | [37] |
| Ipilimumab + Nivolumab | PD-1 and CTLA4 | Recurrent and secondary glioblastoma | Phase II | 37 (not yet recruiting) | N/A | N/A | NCT04145115 | [38] |
| DC vaccines + Nivolumab | CTLA 4 | Recurrent glioblastoma | Phase I | 6 | 15.3 months with surgery | 6.3 months with surgery | NCT02529072 | [39] |
| Oncolytic adenovirus (DNX-2401) + Pembrolizumab | PD-L1 | Recurrent glioblastoma or gliosarcoma | Phase II | 49 (not yet recruiting) | N/A | N/A | NCT02798406 | [40] |
| Stereotactic radiosurgery (SRS) + Spartalizumab + MBG453 | TIM3 + PD-1 + SRS | Recurrent glioblastoma | Phase I | 15 (recruiting) | N/A | N/A | NCT03961971 | [41] |
| Nivolumab + BMS-986205 + Radiotherapy + Temozolomide | IDO + PD-1 | Glioblastoma | Phase I | 30 (recruiting) | N/A | N/A | NCT04047706 | [42] |
| Indoximod + Radiation + Temozolomide | IDO | Pediatric glioblastoma | Phase I | 29 | N/A | 6.2 months | NCT02502708 | [43] |
| Oncolytic Viruses | ||||||||
| Oncolytic virus TG6002 + 5-flucytosine | N/A | Recurrent glioblastoma | Phase I/Phase II | 78 (recruiting) | N/A | N/A | NCT03294486 | [44] |
| Engineered herpes virus G207 | N/A | Recurrent glioblastoma | Phase Ib | 6 | 6.6 months | N/A | NCT00028158 | [45] |
| DNX-2401 + Interferon gamma (IFN-γ) | N/A | Recurrent glioblastoma | Phase Ib | 27 | N/A | N/A | NCT02197169 | [46] |
| Polio/Rhinovirus recombinant (PVSRIPO) | N/A | Recurrent glioblastoma | Phase Ib | 61 | 12.5 months | N/A | NCT01491893 | [47][48] |
| DC Vaccines | ||||||||
| Pembrolizumab + DC vaccine (ATL-DC) | N/A | Recurrent glioblastoma | Phase I | 40 (recruiting) | N/A | N/A | NCT04201873 | [49] |
| Pp65-DCs + Temozolomide | N/A | Glioblastoma | Phase II | 100 (ongoing) | N/A | N/A | NCT02366728 | [50] |
| CAR T Therapy | ||||||||
| Anti-EGFRvIII CAR T cells + Cyclophosphamide | EGFRvIII | Recurrent glioblastoma | Pilot trial | 20 (estimated) | N/A | N/A | NCT02844062 | [51] |
| IL13Ralpha2-specific CAR | IL13Ra2 | Recurrent glioblastoma or refractory high grade glioma | Pilot trial | 3 | 11 months (mean survival) | N/A | NCT00730613 | [52] |
| HER2-CAR VSTs | HER2 | Progressive HER2+ glioblastoma | Phase I | 17 | 11.1 months | N/A | NCT01109095 | [53] |
PD-1, Programmed cell death 1; PD-L1, Programmed cell death ligand 1; CTLA-4, Cytotoxic T lymphocyte protein 4; IDO, Idolamine 2, 3-dioxygenase; CAR T Cells, Chimeric antigen receptors T cells; SRS, Stereotactic radiosurgery; DC vaccines, Dendritic cell vaccines; IFN-γ, Interferon gamma. PVSRIPO, Polio/Rhinovirus Recombinant; ATL-DC, Autologous tumor lysate-pulsed dendritic cell vaccine; Pp65-DCs, Pp65-dendritic cells; Anti-EGFRvIII CAR, anti-epidermal growth factor receptor variant III chimeric antigen receptors; IL13Ralpha2-specific CAR, Interleukin-13 receptor subunit alpha2-specific chimeric antigen receptors. 5-FC, 5-flucytosine; HER2-CAR VSTs, Human epidermal growth factor receptor 2- chimeric antigen receptors virus-specific T cells. OS and PFS were depicted from the time of inoculation. Data taken from https://clinicaltrials.gov/.
3. Novel and Combinatorial Therapies: Preclinical Findings
As the immune-checkpoint blockade strategy becomes more widely available, both synergistic and antagonistic interactions between current standard GBM therapies and ICIs have become a focus of active investigation for better treatment outcome. However, a number of questions remains to be answered, including how to combine different ICIs with conventional therapies (radiotherapy, chemotherapy, and Bevacizumab) and whether the application of combinatorial agents involves positive or negative interactions. In fact, combinatorial GBM treatments will require more careful design for some variables such as mode of delivery, timing, and potential concern for increased toxicity. Moreover, given the expanding number of immunological targets involved in GBM, as well as the extensive list of immunotherapeutic agents under development, the number of available therapeutic combinations is prohibitively large for random testing. Therefore, the rational design of combinational regimens is essential to establish optimal therapeutic strategies. In addition to combinatorial therapy approaches, the focus has been also given to the development of novel therapies such as nanomaterial-based therapy, myeloid cells based strategy, new molecular targeting, and modification of treatment regimens to overcome paramount challenges of GBM treatment with immunotherapy.
3.1. Dual Treatment of PD-1 and TIM3 Blockades with Stereotactic Radiosurgery (SRS)
TIM3 expression in T lymphocytes has been suspected as mechanism of adaptive resistance to anti-PD-1 therapy [54]. In the murine glioma model studies, the upregulation of TIM3 in PD-1 antibody bound T cells was reported after failure of PD-1 blockade treatment [91] and the increase in exhausted PD-1 + TIM3 + T cells was observed in a time-dependent manner with tumor progression [55]. Having determined the co-expression of TIM3 and PD-1 in the T cells, there have been vibrant preclinical investigations on anti-tumor effects of dual PD-1/TIM3 blockade treatment for improved survival [91,92,93][54][55][56]. Following promising preclinical results, a clinical trial to evaluate the combinational use of stereotactic radiosurgery (SRS) with MBG453 (anti-TIM3 antibody) and Spartalizumab (anti-PD-1 antibody) is currently underway (Clinical trial identifier: NCT03961971, Table 1). The rationale of this combination strategy is based on the initial application of local radiotherapy to drive the release of tumor antigens, followed by immunotherapy, to ultimately promote an anti-tumor immune response. SRS has been proven to synergize anti-PD-1 therapy in orthotopic mouse GBM models, by leading to an increase in the amount of CD8+ T cells expressing interferon gamma (IFNγ) as well as a decrease of tumor-infiltrating T reg cells, when compared with a single treatment with SRS or anti-PD-1 therapy [55]. A persistent OS benefit was reported in mice with combinational treatment, and evidence of immune memory was observed by a lack of cancer cell engraftment upon the re-challenge of mice previously treated with anti-PD-1 and SRS [55]. This paradigm has been mainly predicated on the combination of immunotherapy with stereotactic radiosurgery (NCT04225039) [57] as well as laser ablation (NCT02311582) [58], and oncolytic viral treatments (NCT02798406) [40].
3.2. Immunotherapy with Controlled Nano-Drug Delivery System
One major obstacle in GBM therapeutics is that most of the affected patients still present brain regions with an intact BBB, thus restricting drug delivery and greatly compromising the immunological targeting [59][60][61]. One viable route to address this issue is to use local chemotherapy together which harbors synergistic activity with immunotherapy. However, some evidences have indicated that direct and local intracranial delivery could induce several side effects such as infection, edema, and backflow along the catheter. Notably, there has been intense interest in exploiting nanotechnologies that could be applicable for GBM treatment to overcome the constraints of BBB [62][63][64]. Nanotherapeutics combining nanomaterials and GBM targeting molecules not only improve therapeutic efficacy but also circumvent limitations of conventional chemotherapies such as limited permeability, selectivity, and retention [64]. Recently, one research has shown that a mesoporous silica nanoparticle (MSN)-based vehicle coated with interleukin-13 receptor subunit alpha-2 (IL13Rα2)-targeted peptide (IP) using polyethylene glycol (PEG) (namely MSN-PEG-IP, MPI) could be utilized as an effective drug delivery system for GBM therapy. In this work, doxorubicin (DOX)-loaded MPI (MPI/D) has successfully transmitted DOX to GBM cells in vitro and in vivo without affecting normal brain tissues and significantly improve the OS for GBM models [64]. Such significant enhancement on the cellular uptake of DOX in glioma may serve as a potential GBM-targeted drug delivery system [64]. Moreover, another promising finding in this research is that MPI not only delivers DOX to GBM in a targeted manner but also occupies IL13Rα2 and then promotes the binding of IL-13 to IL13Rα1, thus activating the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway to trigger an anti-tumor effect [64].
3.3. A New Perspective on PD-1 Targeted Immune-Checkpoint Inhibitors for Myeloid Cells
Among the array of cancer-related immunotherapies currently available, the immune-checkpoint molecule PD-1 has revolutionized the care of patients with multiple advanced cancers. The PD-1 mAb is believed to interrupt the engagement of PD-1 with its inhibitory ligands, spurring CTL-mediated cancer elimination. It is worth noting that previous work has mainly focused on the anti-tumor role of PD-1 mAb in T cells, but the role of PD-1 mAb in myeloid cells remains unclear. Recently, Strauss and colleagues conducted an exploratory study verifying that the expression of PD-1 in myeloid cells can restrain host immunity against cancers and, therefore, it could be also a good target of anti-PD-1 ICIs [65]. Moreover, researchers generated new mouse models (PD-1f/fLysMcre mice and PD-1f/fCD4cre mice) upon the ablation of PD-1 in myeloid or T cells by conditionally knocking out the respective Pdcd1 alleles. Surprisingly, the inhibition of tumor growth in PD-1f/fLysMcre mice with PD-1 deleted myeloid cells was similar to the one in the mice with complete PD-1 deletion (Pdcd1−/−). Other results have also demonstrated that the anti-tumor efficiency of myeloid-specific deletion of PD-1 (PD-1f/fLysMcre mice) was much higher than the one of T-cell-specific PD-1 ablation (PD-1f/fCD4cre mice) [65]. Myeloid-specific PD-1 ablation induced altered signaling and metabolic reprogramming with enhanced differentiation of effector myeloid cells and emergency myelopoiesis driven synthesis of cholesterol, which is important for the differentiation of inflammatory macrophages and DCs. As a result, the number of immunosuppressive myeloid-derived suppressor cells (MDSCs) was decreased and the amount of differentiated and inflammatory effector myeloid cells was increased. Moreover, the ablation of PD-1 in myeloid cells triggers the increase of effector memory T cells, which have a distinctive function assisting anti-tumor activities. Together, these findings shed new light on the key mechanism of antitumor effect by PD-1 blockade mediated via myeloid cells.
3.4. CD73 Targeting Approach: An Efficient Route to Improve Outcome of Glioblastoma Treatment
Recently, many studies have indicated that the response to ICIs is mostly cancer type dependent. Although the underlying mechanism for those disparities has not been fully understood, current studies have indicated that different immune infiltrates including tumor-infiltrating lymphocytes (TILs) may be correlated with different clinical outcome of specific tumors. More recently, Goswami and colleagues performed comprehensive mass cytometry and single-cell RNA sequencing in patients with NSCLC, prostate cancer, renal cell carcinoma, colorectal cancer, and GBM. The results show that CD73hi macrophages were overexpressed in GBM, which has multiple drug resistance to ICIs. Moreover, MARCO (macrophage receptor with collagenase structure), TGFβ, and multiple SIGLEC (sialic acid-binding immunoglobulin-type lectins) genes are highly expressed on CD73hi cells [66]. According to TCGA (The Cancer Genome Atlas) data, CD73hi gene features are correlated with lower OS rates of GBM patients. Moreover, analysis of GBM samples from patients treated with anti-PD-1 antibodies indicates that this therapy does not cause any significant TME alterations. Such patients still have a high population of CD73hi cells capable of inhibiting T-cell infiltration that lead to poor clinical response to ICIs. Upon knocking out Nt5e (which encodes CD73) in a GBM mouse model, the efficiency of a combinational treatment with anti-CTLA4 and anti–PD-1 was elevated. In particular, the improved efficacy was correlated with obvious enhancement in macrophage polarization toward M1 population and infiltration of T cells in Nt5e knockout mice. Taken together, a variety of evidence has indicated that therapies involving anti-CD73 may function as an efficient approach to improve OS in patients affected by GBM [66].
3.5. Neoadjuvant PD-1 Blockade Treatment to Improve Immunotherapy Efficacy
Despite numerous preclinical successes of anti-PD1 therapy, a recent clinical trial of PD-1 blockade in recurrent GBM has indicated that only 8% patients showed clinically meaningful response [67]. To improve such disappointing therapeutic efficacy, very recent ongoing clinical studies modified treatment regimens by utilizing neoadjuvant checkpoint blockade treatment in addition to standard adjuvant therapy against GBM [68]. Neoadjuvant immunotherapy has been applied in other types of cancers such as lung cancer [69] to boost systemic immunity against tumor antigens, wiping out micro-metastatic cancer deposits that might be the source of postsurgical relapse. In regard to adjuvant treatment that may directly eliminate micro-metastatic tumors upon surgical resection, the application of neoadjuvant PD-1 blockade while the primary tumor is in place may leverage a higher level of endogenous tumor antigen release in the primary tumor to further promote T-cell priming. In particular, immunostimulatory therapy following surgery resection might be more beneficial to reduce residual disease burden and thus improve the likelihood of clinical benefits [69]. To address whether neoadjuvant PD-1 blocking could dramatically change the functional immune landscape and then improve OS in recurrent GBM patients, the Ivy Foundation Early Phase Clinical Trials Consortium has recently conducted a multi-center, open-labeled pilot randomized clinical trial to assess immune response and OS following neoadjuvant as well as adjuvant treatment with pembrolizumab [68]. Respective results have indicated that neoadjuvant PD-1 blocking may significantly down-regulate the expression of genes related to cell cycle but, at the same time, up-regulate the expression of T cell- and interferon-γ-related genes, which are rarely observed in patients who received adjuvant treatment alone. Moreover, decreased PD-1 expression on T cells, reduced monocytic population, the focal induction of PD-1 in TME and strengthened clonal expansion of T cells are frequently observed in neoadjuvant setting when compared with the adjuvant group. Most importantly, treatment with neoadjuvant Pembrolizumab has led to a statistically significant improvement in OS (13.7 months) and PFS (3.3 months), in comparison to the lower OS (7.5 months) and PFS (2.4 months) that was achieved in the adjuvant cohort [68]. Another single-arm phase II clinical trial (Clinical trial identifier: NCT02550249, Table 1) with neoadjuvant nivolumab was conducted [70]. A total of 30 patients involved in this clinical trial received pre- and post-Nivolumab during GBM progression or until reaching intolerable toxicity. As expected, augmented chemokine expression, enhanced T-cell receptor clonal diversity as well as increased immune cell infiltration were observed in a cohort of GBM patients who received the neoadjuvant Nivolumab, thus promoting a local immunomodulatory therapeutic effect [70]. In particular, even though disease relapse was inevitable and no apparent clinical benefits were observed following salvage surgery resection, 2 of 3 GBM patients who received Nivolumab before and after surgery remained disease-free for further 33 and 38 months. These two investigations indicate that neoadjuvant anti-PD-1 therapy may trigger an enhanced local and systemic immune response and significantly improve the median OS and PFS when compared with adjuvant therapy alone, thus acting as a more efficient approach for GBM therapy [70].
Table 2. Management of immune-related adverse events induced by immune-checkpoint inhibitors.
| irAE | ICIs Treatment | Management |
|---|---|---|
| Inflammatory Arthritis | anti-PD-1 and CTLA4 (Ipilimumab/Nivolumab) | Acetaminophen + oral corticosteroids of prednisone + intra-articular corticosteroid injection |
| ICIs resumed | ||
| Temporal Arteritis | anti-PD-L1 (Durvalumab) | Prednisone |
| ICIs resumed pending clinical response | ||
| Myocarditis | anti-PD-1 and CTLA4 (Ipilimumab + Nivolumab) | Methylprednisolone and diuretic treatment |
| ICIs permanently discontinued | ||
| Maculopapular Rash | anti-PD-L1 (Avelumab) | Betamethasone treatment+oral prednisone |
| Resumed after symptom relief below grade 1 toxicity | ||
| Nephritis | anti-PD-1 (Pembrolizumab) | Corticosteroids |
| ICIs resumed after renal indices in a normal range | ||
| Encephalitis | anti-PD-1 and CTLA4 (Ipilimumab + Nivolumab) | Pulse corticosteroids |
| Resumed after neurological recovery | ||
| Myasthenia Gravis | anti-PD-1 (Nivolumab) | Corticosteroids |
| Permanently discontinued | ||
| Uveitus | anti-PD-L1 (Atezolizumab) | Topical cycloplegic agent + prednisone |
| Continued until corticosteroid reduction completed | ||
| Pneumonitis | anti-PD-L1 (Durvalumab) | Dose-dependent corticosteroids according to different grade of checkpoint-inhibitor pneumonitis (CIP) |
| Temporarily held during corticosteroid treatment in grade I and 2 CIP toxicity | ||
| Hypophysitis | anti-PD-1 and CTLA4 (Ipilimumab + Nivolumab) | hydrocortisone +levothyroxine |
| Resumed | ||
| Hypothyroidism | anti-PD-L1 (Durvalumab) | levothyroxine |
| Continued | ||
| Thrombocytopenia | anti-PD-1 (Pembrolizumab) | Prednisone |
| Resumed after 4 weeks treatment | ||
| Colitis | anti-PD-1 and CTLA4 (Ipilimumab + Nivolumab) | Prednisone |
| Resumed |
irAE, Immune-related adverse event; ICIs, Immune checkpoint inhibitors; anti-PD-1, anti-programmed cell death 1; CTLA4, Cytotoxic T lymphocyte protein 4; anti-PD-L1, anti-programmed cell death ligand 1; CIP, Checkpoint-inhibitor pneumonitis.
This entry is adapted from the peer-reviewed paper 10.3390/cancers12092334
References
- Billingham, R.E.; Brent, L.; Medawar, P.B. 'Actively acquired tolerance' of foreign cells. Nature 1953, 172, 603–606, doi:10.1038/172603a0.
- Billingham, R.E.; Brent, L.; Medawar, P.B.; Sparrow, E.M. Quantitative studies on tissue transplantation immunity. I. The survival times of skin homografts exchanged between members of different inbred strains of mice. Proc. R. Soc. Lond. B Biol. Sci. 1954, 143, 43–58, doi:10.1098/rspb.1954.0053.
- Medawar, P.B. Immunity to homologous grafted skin. III. The fate of skin homographs transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br. J. Exp. Pathol. 1948, 29, 58–69.
- Lim, M.; Xia, Y.X.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442, doi:10.1038/s41571-018-0003-5.
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341, doi:10.1038/nature14432.
- Kipnis, J. Multifaceted Interactions Between Adaptive Immunity and the Central Nervous System. Science 2016, 353, 766–771, doi: 10.1126/science.aag2638.
- Bloch, O.; Crane, C.A.; Kaur, R.; Safaee, M.; Rutkowski, M.J.; Parsa, A.T. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res. 2013, 19, 3165–3175, doi:10.1158/1078-0432.CCR-12-3314.
- Wainwright, D.A.; Chang, A.L.; Dey, M.; Balyasnikova, I.V.; Kim, C.K.; Tobias, A.; Cheng, Y.; Kim, J.W.; Qiao, J.; Zhang, L.; et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4 and PD-L1 in mice with brain tumors. Clin. Cancer Res. 2014, 20, 5290–5301, doi:10.1158/1078-0432.CCR-14-0514.
- McGranahan, T.; Li, G.; Nagpal, S. History and current state of immunotherapy in glioma and brain metastasis. Ther. Adv. Med. Oncol. 2017, 9, 347–368, doi:10.1177/1758834017693750.
- Sampson, J.H.; Maus, M.V.; June, C.H. Immunotherapy for brain tumors. J. Clin. Oncol. 2017, 35, 2450–2456, doi:10.1200/JCO.2017.72.8089.
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; de Groot, J.; Li, S.L.; Overwijk, W.W.; et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: Implications for immune checkpoint immunotherapy. Neuro-Oncology 2017, 19, 1047–1057, doi:10.1093/neuonc/nox026.
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J. Clin. Oncol. 2016, 34, 2206–2211, doi:10.1200/JCO.2016.66.6552.
- Johanns, T.M.; Miller, C.A.; Dorward, I.G.; Tsien, C.; Chang, E.; Perry, A.; Uppaluri, R.; Ferguson, C.; Schmidt, R.E.; Dahiya, S.; et al. Immunogenomics of hypermutated glioblastoma: A patient with germline POLE deficiency treated with checkpoint blockade immunotherapy. Cancer Discov. 2016, 6, 1230–1236, doi:10.1158/2159-8290.CD-16-0575.
- Feng, M.; Jiang, W.; Kim, B.Y.S.; Zhang, C.C.; Fu, Y.X.; Weissman, I.L. Phagocytosis Checkpoints as New Targets for Cancer Immunotherapy. Nat Rev Cancer. 2019, 19, 568-586, doi: 10.1038/s41568-019-0183-z.
- Zhao, P.F.; Wang, Y.H.; Kang, X.J.; Wu, A.H.; Yin, W.M.; Tang, Y.S.; Wang, J.Y.; Zhang, M.; Duan, Y.F.; Huang, Y.Z. Dual-targeting biomimetic delivery for anti-glioma activity via remodeling the tumor microenvironment and directing macrophage-mediated immunotherapy. Chem. Sci. 2018, 9, 2674–2689, doi:10.1039/c7sc04853j.
- Li, Q.Y.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242, doi:10.1038/nri.2017.125.
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845, doi:10.1126/science.1194637.
- Ajami, B.; Bennett, J.L.; Krieger, C.; McNagny, K.M.; Rossi, F.M.V. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci. 2011, 14, 1142–1149, doi:10.1038/nn.2887.
- Bennett, F.C.; Bennett, M.L.; Yaqoob, F.; Mulinyawe, S.B.; Grant, G.A.; Hayden Gephart, M.; Plowey, E.D.; Barres, B.A. A combination of ontogeny and CNS environment establishes microglial identity. Neuron 2018, 98, 1170–1183, doi:10.1016/j.neuron.2018.05.014.
- Graeber, M.B.; Scheithauer, B.W.; Kreutzberg, G.W. Microglia in brain tumors. Glia 2002, 40, 252–259, doi:10.1002/glia.10147.
- Matias, D.; Balça-Silva, J.; Da Graça, G.C.; Wanjiru, C.M.; Macharia, L.W.; Nascimento, C.P.; Roque, N.R.; Coelho-Aguiar, J.M.; Pereira, C.M.; Dos Santos, M.F.; et al. Microglia/Astrocytes–glioblastoma crosstalk: Crucial molecular mechanisms and microenvironmental factors. Front. Cell. Neurosci. 2018, 12, 235, doi:10.3389/fncel.2018.00235.
- Hussein, M.R. Tumour-associated macrophages and melanoma tumourigenesis: Integrating the complexity. Int. J. Exp. Pathol. 2006, 87, 163–176, doi:10.1111/j.1365-2613.2006.00478.x.
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272, doi:10.1038/nm.3337.
- Zeiner, P.S.; Preusse, C.; Blank, A.E.; Zachskorn, C.; Baumgarten, P.; Caspary, L.; Braczynski, A.K.; Weissenberger, J.; Bratzke, H.; Reiß, S.; et al. MIF receptor CD74 is restricted to Microglia/Macrophages, associated with a m1‐polarized immune milieu and prolonged patient survival in gliomas. Brain Pathol. 2015, 25, 491–504, doi:10.1111/bpa.12194.
- Pong, W.W.; Walker, J.; Wylie, T.; Magrini, V.; Luo, J.; Emnett, R.J.; Choi, J.; Cooper, M.L.; Griffith, M.; Griffith, O.L.; et al. F11R is a novel monocyte prognostic biomarker for malignant glioma. PLoS ONE 2013, 8, e77571, doi:10.1371/journal.pone.0077571.
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27, doi:10.1038/nn.4185.
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Onco.l 2017, 14, 399–416, doi:10.1038/nrclinonc.2016.217.
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci. Transl. Med. 2017, 9, doi:10.1126/scitranslmed.aal3604
- Tarhini, A.A.; Iqbal, F. CTLA-4 blockade: Therapeutic potential in cancer treatments. Oncotargets Ther. 2010, 3, 15–25, doi:10.2147/ott.s4833
- Wang, H.X.; Xu, T.; Huang, Q.L.; Jin, W.L.; Chen, J.X. Immunotherapy for malignant glioma: Current status and future directions. Trends Pharmacol. Sci. 2020, 41, 123–138, doi:10.1016/j.tips.2019.12.003.
- ClinicalTrials.gov. A Study of the Effectiveness and Safety of Nivolumab Compared to Bevacizumab and of Nivolumab with or without Ipilimumab in Glioblastoma Patients (CheckMate 143). 2013. Available online: https://clinicaltrials.gov/show/NCT02017717 (accessed on 6 June 2020).
- ClinicalTrials.gov. Neoadjuvant Nivolumab in Glioblastoma (Neo-nivo). 2015. Available online: https://clinicaltrials.gov/show/NCT02550249 (accessed on 6 June 2020).
- ClinicalTrials.gov. Nivolumab with Radiation Therapy and Bevacizumab for Recurrent MGMT Methylated Glioblastoma. 2018. Available online: https://clinicaltrials.gov/show/NCT03743662 (accessed on 6 June 2020).
- Reiss, S.N.; Yerram, P.; Modelevsky, L.; Grommes, C. Retrospective review of safety and efficacy of programmed cell death-1 inhibitors in refractory high grade gliomas. J. Immunother. Cancer 2017, 5, 99, doi:10.1186/s40425-017-0302-x.
- ClinicalTrials.gov. Nivolumab and Temozolomide Versus Temozolomide alone in Newly Diagnosed Elderly Patients with GBM (NUTMEG). 2018. Available online: https://clinicaltrials.gov/show/NCT04195139 (accessed on 6 June 2020).
- ClinicalTrials.gov. IL13Ralpha2-Targeted Chimeric Antigen Receptor (CAR) t Cells with or without Nivolumab and Ipilimumab in Treating Patients with Recurrent or Refractory Glioblastoma. 2019. Available online: https://clinicaltrials.gov/show/NCT04003649 (accessed on 6 June 2020).
- ClinicalTrials.gov. Intra-tumoral Ipilimumab Plus Intravenous Nivolumab Following the Resection of Recurrent Glioblastoma (GlitIpNi). 2016. Available online: https://clinicaltrials.gov/show/NCT03233152 (accessed on 6 June 2020).
- ClinicalTrials.gov. A Study Testing the Effect of Immunotherapy (Ipilimumab and Nivolumab) in Patients with Recurrent Glioblastoma with Elevated Mutational Burden. 2020. Available online: https://clinicaltrials.gov/show/NCT04145115 (accessed on 6 June 2020).
- ClinicalTrials.gov. Nivolumab with DC Vaccines for Recurrent Brain Tumors (AVERT). 2016. Available online: https://clinicaltrials.gov/show/NCT02529072 (accessed on 6 June 2020).
- ClinicalTrials.gov. Combination Adenovirus + Pembrolizumab to Trigger Immune Virus Effects (CAPTIVE). 2016. Available online: https://clinicaltrials.gov/show/NCT02798406 (accessed on 6 June 2020).
- ClinicalTrials.gov. Trial of Anti-Tim-3 in Combination with Anti-PD-1 and SRS in Recurrent GBM. 2020. Available online: https://clinicaltrials.gov/show/NCT03961971 (accessed on 6 June 2020).
- ClinicalTrials.gov. Nivolumab, BMS-986205, and Radiation Therapy with or without Temozolomide in Treating Patients with Newly Diagnosed Glioblastoma. 2019. Available online: https://clinicaltrials.gov/show/NCT04047706 (accessed on 6 June 2020).
- ClinicalTrials.gov. Study of the IDO Pathway Inhibitor, Indoximod, and Temozolomide for Pediatric Patients with Progressive Primary Malignant Brain Tumors. 2015. Available online: https://clinicaltrials.gov/show/NCT02502708 (accessed on 6 June 2020).
- ClinicalTrials.gov. Safety and Efficacy of the ONCOlytic VIRus Armed for Local Chemotherapy, TG6002/5-FC, in Recurrent Glioblastoma Patients (ONCOVIRAC). 2017. Available online: https://clinicaltrials.gov/show/NCT03294486 (accessed on 6 June 2020).
- ClinicalTrials.gov. Safety and Effectiveness Study of g207, a Tumor-Killing Virus, in Patients with Recurrent Brain Cancer. 2017. Available online: https://clinicaltrials.gov/show/NCT00028158 (accessed on 6 June 2020).
- ClinicalTrials.gov. DNX-2401 with Interferon Gamma (IFN-γ) for Recurrent Glioblastoma or Gliosarcoma Brain Tumors (TARGET-I). 2014. Available online: https://clinicaltrials.gov/show/NCT02197169 (accessed on 6 June 2020).
- ClinicalTrials.gov. PVSRIPO for Recurrent Glioblastoma (GBM) (PVSRIPO). 2012. Available online: https://clinicaltrials.gov/show/NCT01491893 (accessed on 6 June 2020).
- Desjardins, A.; Gromeier, M.; Herndon, J.E. 2nd.; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. doi: 10.1056/NEJMoa1716435.
- ClinicalTrials.gov. Pembrolizumab and a Vaccine (ATL-DC) for the Treatment of Surgically Accessible Recurrent Glioblastoma. 2020. Available online: https://clinicaltrials.gov/show/NCT04201873 (accessed on 6 June 2020).
- ClinicalTrials.gov. DC Migration Study for Newly-Diagnosed GBM (ELEVATE). 2015. Available online: https://clinicaltrials.gov/show/NCT02366728 (accessed on 6 June 2020).
- ClinicalTrials.gov. Pilot Study of Autologous Anti-EGFRvIII CAR t cells in Recurrent Glioblastoma Multiforme. 2016. Available online: https://clinicaltrials.gov/show/NCT02844062 (accessed on 6 June 2020).
- ClinicalTrials.gov. Cellular Adoptive Immunotherapy Using Genetically Modified T-Lymphocytes in Treating Patients with Recurrent or Refractory High-Grade Malignant Glioma. 2002. Available online: https://clinicaltrials.gov/show/NCT00730613 (accessed on 6 June 2020).
- ClinicalTrials.gov. CMV-Specific Cytotoxic t Lymphocytes Expressing CAR Targeting HER2 in Patients with GBM (HERT-GBM). 2010. Available online: https://clinicaltrials.gov/show/NCT01109095 (accessed on 6 June 2020).
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, e10501, doi:10.1038/ncomms10501.
- Kim, J.E.; Patel, M.A.; Mangraviti, A.; Kim, E.S.; Theodros, D.; Velarde, E.; Liu, A.; Sankey, E.W.; Tam, A.; Xu, H.Y.; et al. Combination therapy with Anti-PD-1, Anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin. Cancer Res. 2017, 23, 124–136, doi:10.1158/1078-0432.CCR-15-1535.
- Sun, F.; Guo, Z.S.; Gregory, A.D.; Shapiro, S.D.; Xiao, G.; Qu, Z. Dual but not single PD-1 or TIM-3 blockade enhances oncolytic virotherapy in refractory lung cancer. J Immunother. Cancer. 2019, 8, e000294, doi:10.1136/jitc-2019-000294.
- Patel, M.A.; Kim, J.E.; Theodros, D.; Tam, A.; Velarde, E.; Kochel, C.M.; Francica, B.; Nirschl, T.R.; Ghasemzadeh, A.; Mathios, D.; et al. Agonist anti-GITR monoclonal antibody and stereotactic radiation induce immune-mediated survival advantage in murine intracranial glioma. J. Immunother. Cancer 2016, 4, e28, doi:10.1186/s40425-016-0132-2.
- Washington University School of Medicine; Sharp, M.; Corp, D. MK-3475 in Combination with MRI-Guided Laser Ablation in Recurrent Malignant Gliomas. Available online: https://clinicaltrials.gov/ct2/show/NCT02311582 (accessed on 15 July 2020).
- Pardridge, W.M. Blood–brain barrier delivery. Drug Discov. Today 2007, 12, 54–61, doi:10.1016/j.drudis.2006.10.013.
- Weiss, N.; Miller, F.; Cazaubon, S.; Couraud, P. The blood-brain barrier in brain homeostasis and neurological diseases. Biochim. et Biophys. Acta (BBA)-Biomembr. 2009, 1788, 842–857, doi:10.1016/j.bbamem.2008.10.022.
- Da Ros, M.; De Gregorio, V.; Iorio, A.L.; Giunti, L.; Guidi, M.; De Martino, M.; Genitori, L.; Sardi, I. Glioblastoma chemoresistance: The double play by microenvironment and blood-brain barrier. Int. J. Mol. Sci. 2018, 19, e2879, doi:10.3390/ijms19102879.
- Liu, Y.Y.; Ran, R.; Chen, J.T.; Kuang, Q.F.; Tang, J.; Mei, L.; Zhang, Q.Y.; Gao, H.L.; Zhang, Z.R.; He, Q. Paclitaxel loaded liposomes decorated with a multifunctional tandem peptide for glioma targeting. Biomaterials 2014, 35, 4835–4847, doi:10.1016/j.biomaterials.2014.02.031.
- Shi, J.L.; Hou, S.Q.; Huang, J.F.; Wang, S.S.; Huan, W.; Huang, C.J.; Liu, X.J.; Jiang, R.; Qian, W.B.; Lu, J.J.; et al. An MSN-PEG-IP drug delivery system and IL13Rα\alphaα2 as targeted therapy for glioma. Nanoscale 2017, 9, 8970–8981, doi:10.1039/c6nr08786h.
- Strauss, L.; Mahmoud, M.A.A.; Weaver, J.D.; Tijaro-Ovalle, N.M.; Christofides, A.; Wang, Q.; Pal, R.; Yuan, M.; Asara, J.; Patsoukis, N.; et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020, 5, doi:10.1126/sciimmunol.aay1863.
- Goswami, S.; Walle, T.; Cornish, A.E.; Basu, S.; Anandhan, S.; Fernandez, I.; Vence, L.; Blando, J.; Zhao, H.; Yadav, S.S.; et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat. Med. 2020, 26, 39–46, doi:10.1038/s41591-019-0694-x
- Goswami, S.; Walle, T.; Cornish, A.E.; Basu, S.; Anandhan, S.; Fernandez, I.; Vence, L.; Blando, J.; Zhao, H.; Yadav, S.S.; et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat. Med. 2020, 26, 39–46, doi:10.1038/s41591-019-0694-x
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent glioma clinical trial, CheckMate-143: The game is not over yet. Oncotarget 2017, 8, e91779, doi:10.18632/oncotarget.21586.
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486, doi:10.1038/s41591-018-0337-7.
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N. Engl. J. Med. 2018, 378, 1976–1986, doi:10.1056/NEJMoa1716078.
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; de Andrea, C.; López-Diaz De Cerio, A.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476, doi:10.1038/s41591-018-0339-5.
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; de Andrea, C.; López-Diaz De Cerio, A.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476, doi:10.1038/s41591-018-0339-5.