The carotid body (CB) is a vital sensory organ, located near the carotid bifurcation, that constantly monitors blood supplying the brain. The CB is stimulated by acute hypoxia, upon which it rapidly activates vital cardiovascular and respiratory reflexes, including peripheral vasoconstriction, elevated heart rate and increased breathing. These ensure that sufficient blood-oxygen is delivered to the brain to support survival. It is now apparent that there are numerous G-protein-coupled receptors (GPCRs) expressed in the CB, and that modulation of these receptors is able to alter baseline CB activity and the sensitivity to hypoxia. Indeed, components of GPCR signaling pathways represent some of the most highly expressed genes in the rodent CB.
- G-protein
- GPCR
- carotid body
- hypoxia
- hypertension
- drug-discovery
1. Gαs and Gαi Protein-Coupled Receptor Signaling in the Carotid Body
1.1. Adenosine and CD73
Adenosine is a precursor and a breakdown product of an important excitatory neurotransmitter in the CB, ATP, which is tonically released from the type I cell [1]. Extracellular concentrations of adenosine and ATP are reported to be approximately 20 pmol/CB and 4 pmol/CB respectively, in normoxia and increase by 174% and 147% in mild hypoxia in the rat CB [2][3]. A2A and A2B-receptor mRNA has been isolated from rat CBs, and immunocytochemical and in situ hybridization techniques have shown that A2A- and A2B-receptors are present on the type I cell and A2A-receptors are expressed on post-synaptic sensory fibers [4][5][6][7]. In contrast, A1- and A3-receptors do not appear to be present in the CB [4][7].
CB activation by exogenous adenosine in vivo evokes an acute increase in respiratory frequency, tidal volume and minute ventilation in the rat[8]. More recent studies have now also confirmed that acute adenosine administration elevates ventilation in humans[9]. Exogenous adenosine is capable of activating all aspects of the CB chemotransduction cascade, including inhibition of Twik-related acid-sensitive K+ (TASK) and voltage-gated K+ channels, elevation of [Ca2+]i, promotion of neurotransmitter release and stimulation of chemoafferent fibers [6][10][11][12].
Endogenous adenosine plays an important role in establishing baseline neurotransmitter release and chemoafferent activity in normoxia [6][13]. The non-selective A2-receptor antagonist 8-(p-Sulfophenyl)theophylline (8-SPT) causes more than 90% depletion in chemoafferent frequency when measured in the ex vivo CB preparation [13]. This is particularly important given that it is the rise in baseline chemoafferent activity that most likely promotes chronic reflex stimulation and neurogenic hypertension associated with OSA, SH and HF. Initial reports suggest that caffeine (an adenosine receptor antagonist), does not modify the basal chemoafferent frequency in animals following exposure to chronic intermittent hypoxia (CIH), a robust model of OSA [14]. However, it must be noted that these animals displayed a reduction rather than elevation in baseline nerve activity, which is not characteristic of other studies using CIH. More experiments are definitely warranted to evaluate this further as well as a potential role for adenosine in mediating CB hyperactivity in SH and HF.
Adenosine also has an important role in mediating CB responses to hypoxia [15][16][17] and hypercapnia [13][18]. Inhibition of adenosine receptors has a greater impact at low-intensity levels of hypoxia [3], suggesting that adenosine is important in establishing CB hypoxic sensitivity and/or that adenosine release predominates under milder hypoxic conditions. However, the exact signaling mechanism remains controversial. Both A2A- and A2B-receptors are coupled by the Gαs protein, thus a role for adenylyl cyclase (AC) activation and cAMP accumulation appear highly likely [1][19]. In the whole CB preparation, A2B-, but not A2A-receptor antagonists depress catecholamine secretion in hypoxia [6][20]. In these same studies, it was reported that both A2A- and A2B-receptor antagonists reduce chemoafferent frequency in hypoxia, leading the authors to conclude that adenosine acts on pre-synaptic A2B- and post synaptic A2A-receptors. This is supported by the finding that A2B- not A2A-receptor inhibition suppresses type I cell catecholamine secretion and rises in [Ca2+]i induced by adenosine in co-cultures with petrosal neurons [5]. However, in an earlier study, it was observed that A2A-receptor antagonism almost completely abolished the adenosine-mediated elevation in Ca2+, an effect that was mimicked by the protein kinase A (PKA) inhibitor H89 [12]. Other studies have shown that H89 and other PKA inhibitors do not significantly alter the catecholamine release caused by hypoxia [21]. This has led to the hypothesis that whilst increased adenosine and cAMP are important, they may be acting independently of PKA. Alternative messengers of cAMP include exchange proteins activated by cAMP (EPACs), which when activated, can overcome the inhibition of AC in the CB [21]. EPAC is suggested to be a key regulator of exocytotic machinery and K+ channels. Although no direct link between adenosine and EPAC has yet been established, as studies so far have only looked at the link between hypoxia and EPAC, a link is plausible and could be the focus of future investigations. Another target could be the hyperpolarization-activated cation current Ih reported to be activated via an A2A-receptor and cAMP-dependent mechanism on the post-synaptic site [22].
An important consideration is the actual source of adenosine in normoxia and hypoxia. It has been suggested that adenosine may be formed inside type I cells by the breakdown of cAMP and ATP and subsequently released through the bidirectional equilibrative nucleoside transporter (ENT) into the synapse [2][3]. This source of adenosine might be expected to increase proportionally during hypoxia as oxidative phosphorylation decreases and cAMP increases. It is therefore somewhat surprising that the functional impact of adenosine is more apparent at low- rather than high-intensity hypoxia [3]. Furthermore, our own experiments found that pharmacological inhibition of the ENT did not modify chemoafferent frequency in either normoxia or hypoxia [17]. This is possibly suggestive of an alternative source of adenosine.
It is now apparent that adenosine can be generated from extracellular ATP (Figure 1). In the CB, a significant synaptic ATP concentration is due to the tonic vesicular neurosecretion from the type I cell [23] and also through ATP release from type II cells via pannexin-1 channels [24]. There may also be ATP release from red blood cells, although this remains to be clarified in the CB circulation. CD73 (ecto-5′-nucleotidase) catalyzes the formation of adenosine from AMP, following initial breakdown of ATP and adenosine diphosphate (ADP) by CD39 (ecto-nucleosidetriphosphate diphosphohydrolase) [25]. RNA and protein expression of CD73 have now been identified in the CB with the majority being localized to the type I cell [26]. Inhibiting CD73 with α,β-Methylene-ADP (AOPCP) decreases the total pool of extracellular adenosine in the CB [2]. Furthermore, similar pharmacological inhibition of CD73 causes a dramatic reduction in basal chemoafferent activity and blunts the CB response to hypoxia in vitro and the cardiovascular-respiratory response to hypoxia in vivo [17]. Given the high concentrations of AOPCP used in these studies, there is still a need to validate these findings using genetic models or with lower doses of more selective CD73 inhibitors that are becoming available. Consistent with a hypoxia-inducible factor-1 (HIF-1)-dependent elevation of CD73 in other tissues, including hypoxic tumors [27], CB CD73 expression increases in response to chronic hypoxia along with A2B-receptors [5][28]. Interestingly, a recent study shows that CD73 expression is elevated in the aged CB, despite an overall reduction in chemoreceptor function[29]. However, a role for CD73 in mediating CB hyperactivity in CIH, SH or HF remains to be elucidated.
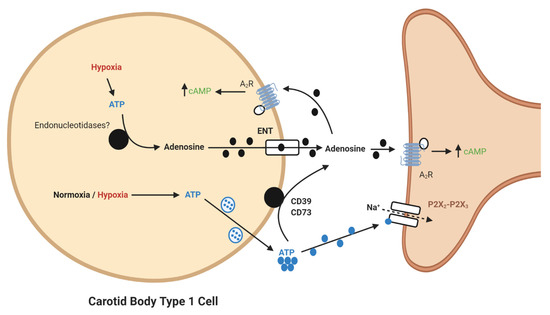
Figure 1. Schematic illustration of ecto-5′-nucleotidase (CD73)-mediated adenosine generation and signaling in the carotid body (CB). During normoxia/hypoxia, ATP released as a neurotransmitter can be converted to adenosine by the action of ecto-nucleosidetriphosphate diphosphohydrolase (CD39) and CD73. Alternatively, ATP can be converted to adenosine in the type I cell and released via the equilibrative nucleoside transporter (ENT). Adenosine binds to A2-receptors on the pre- and post-synaptic membrane to increase baseline activity and overall hypoxic sensitivity. Filled lines denote purinergic signaling, dashed lines denote ion flow.
1.2. Adrenaline
Although adenosine is regarded as the major positive regulator of cAMP in the CB, there are other substances that may elevate cAMP. One of these is adrenaline. Adrenaline binds to β-adrenoceptors, most commonly the β1 and β2 subtypes which are coupled to the Gαs subunit, resulting in cAMP elevation in heart and other tissue [30]. Although there is substantial evidence that adrenaline stimulates breathing [31][32], the direct effect on the CB is less clear. There are reports of exogenous adrenaline administration in vivo causing both increased and decreased CB chemoreceptor discharge [31][33][34]. The effect might be dependent on the dose/concentration used, whilst physiological concentrations selectively act on β-adrenoceptors, supra-physiological levels could act on D2-receptors leading to inhibition. This is supported in our own work where we identified a stimulatory effect at 10 nM but an inhibitory action at higher concentrations, as evidenced by a marked reduction in chemoafferent frequency [32][35].
What is more important to consider is the physiological relevance of endogenous adrenaline. Our own understanding of the effects of endogenous adrenaline have been developed through the study of the counter-regulatory response to hypoglycemia. During hypoglycemia, adrenaline levels rise [36], and hepatic glucose release increases to counter this fall. Multiple studies have now provided evidence of activation of the CB in response to hypoglycemia in animals [37][38] and humans [39][40]. This is essential not only to further augment hepatic glucose release but also to increase ventilation to match the concurrent rise in metabolic rate, initiated by adrenaline [38][41](Figure 2). In these circumstances, a heightened CB CO2 sensitivity allows for increased chemoreceptor discharge and ventilation in the absence of any change in arterial blood gases. Recent evidence has shown that the increase in minute ventilation and CO2 sensitivity during hypoglycemia was blunted by propranolol treatment, removal of the adrenal gland and section of the carotid sinus nerve (CSN) [32]. This is consistent with the view that it is adrenaline that activates the CB during hypoglycemia, via β-adrenoceptors. Key experiments are now needed to see if this is also evident in humans. Perhaps surprisingly, no studies have yet looked at a potential role for adrenaline in causing CB hyperactivity in OSA or HF, both of which are associated with a chronic rise in plasma catecholamines. Chronic isoprenaline delivered via osmotic mini-pump does elevate baseline breathing and responses to hypoxia and hypercapnia [35], but as yet, the relevance of this model to pathology remains uncertain and studies in animals and/or humans with HF and CIH are required [42]. Moreover, there is hardly any indication of how adrenaline acts to augment CB chemosensitivity (even the specific receptor subtypes) and there is limited evidence of the impact of beta-blockers on CB function. Key experiments are needed to establish if chronic beta-blocker treatment (which are widely prescribed for heart failure, hypertension and arrhythmia) dampens CB function such that there is increased vulnerability to hypoxic and hypoglycemic exposure. As such, we are just beginning to grasp the importance of adrenaline in mediating CB function in health and disease.
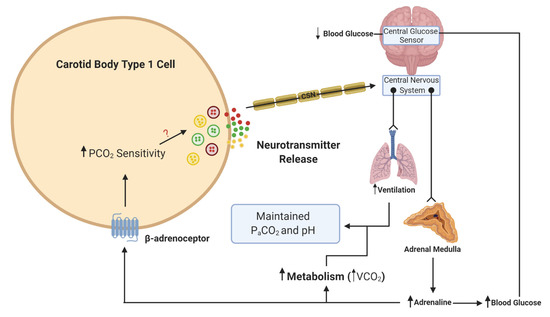
Figure 2. Schematic illustration of adrenaline activation of the carotid body (CB) during hypoglycemia. When blood glucose decreases, this is sensed in the brainstem and leads to a reflex increase in adrenaline release from the adrenal medulla. β-adrenoceptors in type 1 cells will be activated by adrenaline. This activation will lead to an increase in CO2 sensitivity, neurotransmitter release and an increase in ventilation. The elevation in ventilation matches the increase in metabolic rate and CO2 generation (VCO2), such that the overall partial pressure of arterial CO2 (PaCO2) and pH remain constant. The ? denotes that the signaling mechanism linking increased CO2 sensitivity with enhanced neurotransmitter release is still unknown. Lines with arrows denote signaling pathways. Lines with circles and chevrons denote efferent neurons.
1.3. Lactate and Olfr78
Lactate signaling through the GPCR Olfr78 (Gαs) has gained significant attention based on the finding that Olfr78 mRNA is highly expressed in the murine CB and its global deletion completely abolishes the hypoxic ventilatory response (HVR) [43]. This was coupled with a complete depression of CB chemoafferent activity during hypoxia. Indeed, it is logical that both intracellular and systemic lactate levels increase during hypoxia after partial or complete termination of oxidative phosphorylation in the CB and other tissues, thereby providing a stimulus for Olfr78. The work has now been strengthened by evidence from another laboratory demonstrating a similar depression in the HVR in a different mouse strain [44]. However, this was in spite of severe hypoxia failing to depress the elevation in type I cell [Ca2+]i in Olfr78−/−, raising questions over the exact location of the Olfr78 signaling within the CB or chemoreflex pathway. Furthermore, this same study observed similar elevations in chemoafferent nerve activity induced by lactate in Olfr78−/− and wildtype (WT) CBs, suggesting that another ligand, and not lactate, activates Olfr78 during hypoxia. In contrast to these studies, it has also been reported that that Olfr78−/− mice maintain a robust HVR and retain type I cell sensitivity to hypoxia [45]. Clearly there is a need to reconcile these findings and in particular to study lactate and Olfr78 in higher species, including humans, which have a larger chemoafferent response to hypoxia and are less susceptible to respiratory alkalosis. In our own studies, we observed that after prolonged exposure to glucose deprivation, which eventually caused chemoafferent excitation, 5 mM lactate acted to suppress rather than excite chemoafferent frequency [46]. Despite being in a different experimental setting, we propose that lactate may have an additional role as an alternative energy source, independent of Olfr78, similar to that seen in the central nervous system (CNS) and peripheral nerves [47][48].
1.4. Dopamine and Noradrenaline
The CB contains a large amount of stored catecholamines (CAs) in dense core vesicles, and for its mass, the overall CA content is equivalent only to that seen in adrenal medullary tissue [49]. The enzyme tyrosine hydroxylase (TH), that initiates the synthesis of CAs from tyrosine, has now emerged as a well-established type I cell marker [50]. Dopamine (DA) is the most abundant CA in type I cells, forming more than 50% of the overall CA content [49][51]. There is some evidence of dopamine beta-hydroxylase expression in type I cells, consistent with small amounts of noradrenaline (NA) [52]. Interestingly, dopamine beta-hydroxylase is augmented in CBs of SH rats, possibly indicative of a more pronounced role of NA as a transmitter in pathology [53]. Recently, it has been demonstrated that type I cells contain significant amounts of vesicular monoamine transporter 1 (VMAT1), the enzyme most likely responsible for incorporation of DA and NA into secretory vesicles [54][55]. This process may also be highly sensitive to levels of biotin, a vitamin and coenzyme known to accumulate in type I cells [54]. Future experiments may well evaluate how these enzymes are altered in CB-mediated cardiovascular and metabolic disease.
DA is released from type I cells in abundance during hypoxia [56][57], but its action seems to be autoinhibitory, providing the CB with a degree of inhibitory feedback control via D2-receptors [58][59][60][61]. Indeed, it has been suggested that the balance between dopamine (via Gαi-coupled D2-receptors) and adenosine (via Gαs-coupled A2B-receptors) is critical in determining the overall cAMP level and excitability within the type I cell [20]. The increase in [Ca2+]i in rat type I cells in response to hypoxia can be attenuated by D2-receptor agonists [62]. D2-receptors have now also been positively confirmed in the human CB [63] and DA infusion to depress CB function is commonly used to estimate CB contribution to pathophysiological reflexes in humans [64][65][66]. Whether or not DA completely silences the CB chemoafferent discharge in humans is unknown. Exogenous DA does indeed reduce (but not abolish) the HVR in humans, but there is some considerable variability between individuals and not all of the observed effects may be solely attributable to CB inhibition [67][68]. In mice with global deficiency of D2-receptors, whilst type I cell neurotransmitter release was enhanced in response to hypoxia, the chemoafferent activity was reduced and the HVR was unchanged [69]. In rats, the HVR can be enhanced rather than depressed, by systemic inhibition of the monoamine oxidase enzyme, which is likely to have increased free DA [70]. These studies are suggestive of additional locations and possible excitatory functions of D2-receptors within the CB and/or chemoreflex pathway other than the type I cell in rodents. Any excitatory action does not however seem to involve cAMP sensitive hyperpolarization-activated currents (Ih), as evidenced by DA causing a reduction in this post-synaptic current through a D2-receptor-mediated mechanism [22].
Reverse transcription polymerase chain reaction (RT-PCR) analysis of short- and long-term hypoxic rats’ CBs showed a time-dependent increase in the expression of TH and D2-receptor genes [71][72]. After 48 hours of hypoxia, D2-receptor mRNA levels decreased, but after 7 days, the expression of D2-receptor increased significantly [71]. The alteration in D2-receptor expression has been hypothesized to cause a change in DA signaling in the CB which contributes to the changes in ventilatory adaptation observed with long-term hypoxia associated with chronic obstructive pulmonary disease (COPD) or HF [73]. A role for DA in establishing CB hyperactivity in response to CIH has recently been investigated [74]. In this study, CIH augmented CB DA content, CB catecholamine release and arterial blood pressure, but not the HVR. Moreover, the D2-receptor antagonist domperidone reversed the elevation in blood pressure and the excessive catecholamine release during hypoxia in CBs isolated from CIH animals. This again goes against the idea that DA is simply an inhibitory neurotransmitter and opens up the possibility that it has an excitatory function in the CB in certain pathological conditions. In this instance, the authors do also point out that some of the effects of domperidone may have been due to changes in either systemic or local CB blood flow.
NA makes up approximately 15–40% of the total catecholamine content of the CB (varying in different species) and is located in the dense core secretory vesicles in type I cells [75][76]. In acute hypoxia, NA is secreted from the type I cells into the extracellular space in proportion to the stimulus intensity [75]. Release of NA from sympathetic terminals and uptake of NA from the circulation also contributes to the total extracellular NA content in the CB. Despite its concentration being significantly lower than that of DA, NA still has an important functional role in CB chemoreception. In a study performed in anaesthetized dogs in the 1970s, it was first observed that infusion of NA into the carotid artery produced a burst of chemoafferent excitation followed by a more sustained inhibition [77]. Similar intracarotid infusions of α2-adrenoceptor agonists produced chemoafferent inhibition and blunted the response to hypoxia in anaesthetized cats [78]. Intracarotid injection of NA has been shown to inhibit ventilation in goats, a response that was attenuated by both α-adrenoceptor and D2-receptor antagonists [79]. Sectioning of sympathetic nerves innervating the CB in cats was shown to have no effect on baseline chemoafferent activity but did augment the frequency during hypoxia [80]. Direct application of NA to the whole ex vivo CB or dissociated type I cells blunts neurotransmitter release, Ca2+ currents and the rise in cAMP during hypoxia, effects which are again dependent on α2-adrenoceptor activation [81][82]. Thus, there is a strong body of evidence to suggest that endogenous NA released from sympathetic nerves and type I cells promotes inhibitory feedback via Gαi-coupled α2-adrenoceptors. In contrast, we have seen that venous infusion of NA in anaesthetized rats elevates ventilation that is partially blocked by section of the CSN [35]. It is possible that this method of administration produced a more sustained/intense vasoconstriction to arterioles supplying the CB, leading to a significant reduction in blood flow and local hypoxia. Alternatively, wide-spread delivery of NA could promote the release of intermediate substances into the systemic circulation that act indirectly on the CB to evoke chemostimulation. Clearly, there is a need to unify these contradictory findings. The impact of sympathetic stimulation on CB blood flow requires more investigation and especially in conditions where there is a known increase in sympathetic tone in other vascular beds. Furthermore, it is currently unknown what the chronic effect of elevated plasma NA could be on CB blood flow and function, especially common in conditions such as OSA, SH, HF and pheochromocytoma.
2. Gαq-Protein-Coupled Receptor Signaling in the Carotid Body
2.1. Angiotensin II
Angiotensin II (Ang II) is a powerful hormone that influences many organs and its level is increased in important cardiovascular diseases including HF, hypertension and chronic kidney disease. A wealth of evidence has also shown that Ang II acutely increases CB chemoafferent frequency with a threshold in the pico/nano-molar range [83][84]. Rises in [Ca2+]i initiated by Ang II can be attenuated by losartan, an angiotensin 1 (AT1)-receptor antagonist [85]. RT-PCR analysis revealed that both AT1a and AT1b subtypes are expressed in type I cells and immunohistochemistry has confirmed the presence of AT1-receptor protein [85][86]. Combined with the finding that an AT2-receptor antagonist was not able to prevent the Ang II-induced increase in [Ca2+]i, these results suggest that AT1-receptors are responsible for modifying CB activity via Ang II. AT1-receptors are coupled to the Gαq protein and its activation leads to the generation of IP3, via phospholipase C, causing the release of Ca2+ from intracellular stores [87]. It is now thought that this rise in Ca2+ is sufficient to activate the non-selective cation channel which contributes to cellular depolarization [88]. Repetitive application of Ang II can also promote sensory long-term facilitation (sLTF) of chemoafferent nerve activity, an effect reliant on activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and ROS generation [84]. AT1-receptor signaling is also key to causing CB sensory long-term facilitation (sLTF) and persistent sympathoexcitation following acute intermittent hypoxia, in a protein kinase C (PKC), ROS-dependent manner [89][90]. Whether or not the response to prolonged Ang II desensitizes due to receptor phosphorylation, β-arrestin recruitment and internalization is yet to be studied in the CB. Recent reports have indicated that AT1-receptor inhibition does not reduce the HVR or chemoreflex in healthy humans before or after a short period (8 h) of hypoxia [91]. Furthermore, raising serum Ang II in healthy individuals does not lead to chemoreflex sensitization [92]. This possibly indicates that Ang II and AT1-receptor signaling might be more important in pathology or short/long-term CB adaptation rather than setting chemosensitivity in healthy individuals.
Chronic hypoxia has been shown to increase AT1a and AT1b-receptor mRNA expression in type I cells as well as CSN sensitivity to Ang II [93]. In situ hybridization, PCR and Western blot analysis have confirmed the presence of components of the renin-angiotensin system (RAS), including angiotensinogen and angiotensin-converting enzyme (ACE), in type I cells of the CB, indicative of a local system [94]. These components are elevated in rat CBs exposed to chronic hypoxia and CIH [94][95]. AT1-receptor protein is also increased in CBs following CIH [96]. It has therefore been suggested that Ang II may play a role in causing CB hyperactivity in diseases associated with chronic hypoxia and/or CIH, e.g., in OSA. To support this, heightened sympathetic activity following exposure to CIH is prevented in rats treated with losartan, an AT1-receptor inhibitor [96]. Translational findings in humans are currently limited. However, in a recent clinical trial, it was observed that losartan reduced systolic and diastolic blood pressure in OSA patients without modifying muscle-sympathetic outflow or ventilation during hypoxia [97]. OSA was fairly well established in these patients and as such, there may have been irreversible epigenetic remodeling of CB function [98], accounting for the apparent lack of impact of losartan on CB function. Future studies could evaluate if losartan or similar agents are more effective in protecting against the development of hypertension in newly diagnosed or more-mild OSA patients.
In animal models of HF, augmented CB Ang II signaling is also suggested to increase basal and hypoxic sympathetic outflow and contribute to neurogenic hypertension [99]. AT1-receptor antagonists reduce chemoafferent firing frequency and renal sympathetic nerve activity in rabbits with HF but not controls [99]. Furthermore, The AT1-receptor antagonist, L-158,809 (1 μM), significantly decreases the sensitivity of voltage-dependent K+ current to hypoxia in type I cells isolated from CH but not control rabbits [100]. These increased actions of Ang II in HF are reported to be mediated by increased NADPH-oxidase activity and ROS generation [101]. Clinical trials are now called for to directly assess the role of Ang II and Ang II antagonists in mediating CB hyperactivity and hypertension in human patients with HF.
3.2. Serotonin (5-HT)
Serotonin (5-HT) is another excitatory neurotransmitter in the CB and the enzymes involved in its biosynthesis (tryptophan hydroxylase) and transport (5-HT transporters) have been identified in type I cells, indicating that 5-HT is synthesized locally [102][103]. During normoxia, the release of 5-HT occurs spontaneously from type I cells in large clusters (ca 20 cells) to establish baseline spike-like depolarizations [104][105]. Exogenous 5-HT evokes a significant increase in [Ca2+]i in a distinct population of type I cells [106]. 5-HT binds to the Gαq-linked 5-HT2A-receptor subtype expressed in type I cells and the stimulatory effect is via a PKC-mediated inhibition of the Ca2+-activated K+ current [110]. Hypoxia induces depolarization of type I cells leading to neurotransmitter release, including 5-HT. 5-HT2-receptor antagonists reduce both the number and magnitude of type I cell spike depolarizations and the hypoxia-stimulated rise in [Ca2+]i [107]. However, nerve-recording studies have demonstrated that 5-HT does not significantly alter the peak response to hypoxia, but does prolong the duration of the response [108]. 5-HT has also been implicated in contributing to chemoafferent hyperactivity in the CB during pathophysiological conditions associated with CIH [109]. 5-HT release and 5-HT2-receptor activation are crucial for NADPH oxidase (NOX) activation during CIH [109]. Hypoxia induces a significant increase in 5-HT release from type I cells in CIH-treated animals, which in turn leads to an increase in PKC activity and NADPH-oxidase 2-derived ROS generation, which is suggested to cause persistent chemoafferent hyperactivity [110]. Taken together, these results suggest a role for 5-HT in setting CB excitability and in both normal and pathological conditions. However, as with Ang II, it is not known whether the response to prolonged 5-HT desensitizes and if some of the longer-lasting effects of 5-HT in pathology are dependent on β-arrestins. In addition, selective targeting of the CB with 5-HT2-receptor antagonists is yet to be trialed in humans with OSA.
3. Receptors with Dual Gαs/Gαq Signaling
3.1. Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP) and the PAC1-Receptor
Pituitary adenylate cyclase-activating polypeptide (PACAP) is a neuropeptide that was originally identified in the hypothalamus and was observed to be capable of increasing cAMP content in pituitary cell cultures [111]. However, since PACAP can be detected in peripheral blood [112], there is also the potential for systemic actions, including on the CB. PACAP was initially suggested to exert an excitatory influence on the type I cell by inhibiting background TASK currents and raising [Ca2+]i [113]. This is now thought to occur via activation of pituitary adenylate cyclase-activating polypeptide type 1 (PAC1)-receptors. Interestingly, the activation of PAC1-receptors can lead to stimulation of both Gαs and Gαq signaling pathways. In experiments utilizing the ex vivo intact CB preparation, it was shown that prolonged PACAP application (50 min, 200 nM) caused a biphasic response characterized by an initial short-lasting peak 2–3-fold elevation in chemoafferent discharge, followed by a drop and plateau at approximately 1.5–2-fold above baseline [114]. Whilst inhibitors of PKA and EPAC did inhibit chemoafferent responses, these were modest compared to the effect of the PLC inhibitor U73122, which strongly suppressed both the peak and steady-state responses [114]. Thus, although there are roles for both Gαs and Gαq, it is more likely that in this case, Gαq predominates.
Of particular importance is the potential link between PACAP and sudden infant death syndrome (SIDS) in African Americans [115]. In this pilot study, a significant association was observed between SIDS and a single nucleotide polymorphism in exon 2 of the PACAP gene, but only in African American infants and not Caucasians [115]. The reason for the observed differences is still to be elucidated. Further studies are required to explore this relationship in much larger patient cohorts and in different ethnicities. However, it is known that neonatal animals that lack the PACAP gene have high mortality due to significantly reduced respiratory responses to both hypoxia and hypercapnia, and a high incidence of prolonged apneas [116]. Furthermore, neonatal mice lacking the PAC1-receptor have a reduced HVR compared to WT littermates [117]. Administration of PACAP-38, an exogenous form of PACAP, in the rat in situ working heart-brainstem preparation induced a significant increase in respiratory frequency, an effect that was abolished by CB denervation [118]. PACAP-38 also stabilized breathing in this preparation, however, this was not modified by CB denervation, indicative of a central, CB-independent action. Thus, the role for PACAP and the CB in SIDS is still to be defined, and there is an urgent need to identify more personalized protective therapies for vulnerable infants at particularly high risk.
4.2. Endothelin
Endothelin (ET) is a 21 amino acid peptide most commonly known for its ability to cause potent vasoconstriction. The G-protein-coupled receptors ETA and ETB are expressed in the CB type 1 cells as well as in the surrounding blood vessels [119]. ETA- and ETB-receptors can be coupled to multiple G-proteins, including Gαq, Gαs and Gαi,. Given that ET causes type I cell stimulation and an increase in [Ca2+]i, they are most likely to be linked with Gαq and Gαs in this tissue [120]. The main function of ET signaling appears to be in mediating CB adaptation to chronic hypoxia and CIH in adults [121][122] and neonates [123]. In adult rats, chronic hypoxia upregulates ETA- and ETB-receptor expression in the type I cell and ETA-selective antagonists have a strong impact on chemoafferent discharge, producing ca 11% inhibition before chronic hypoxia, increasing to ca 50% inhibition after 16 days of chronic hypoxia [124][125]. In chronic hypoxia, increased endothelin signaling seems to overlap with enhanced generation of nitric oxide (NO), a substance capable of both increasing and decreasing CB chemosensitivity dependent on the specific microdomain in which it is produced [124][126]. Further work is necessary to define the exact role and location of this increased NO generation in chronic hypoxia. In contrast, exposure to CIH appears to increase expression of ETB but not ETA [119]. Non-selective ET-receptor antagonists have a greater inhibitory effect on chemoafferent activity in CIH compared to control rat and cat CBs [127][128]. In a recent study, it was suggested that the augmented ET signaling includes upregulation of PLC and PKC [129]. In neonatal rat CBs, the elevation in chemoafferent activity is dependent on ETA-receptor stimulation and ROS generation [124]. These important findings can be advanced further by evaluating ET signaling and CB hyperactivity in humans and in other CB-mediated pathology, such as HF and spontaneous/essential hypertension. A summary of G-protein signaling in the CB type I cell is presented in Figure 3.
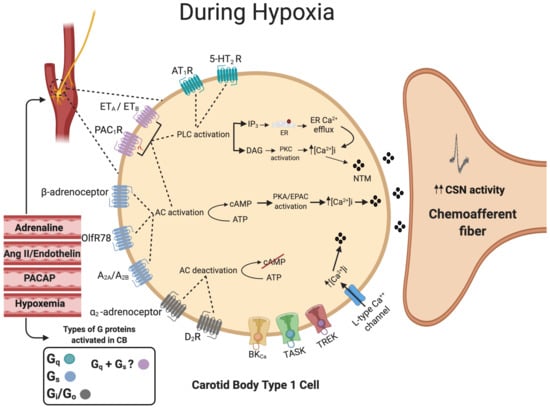
Figure 3. Summary of G-protein signaling in the carotid body type I cell. The activation of Gq-protein-coupled receptors (Green) activates phospholipase C (PLC), which in turn leads to the formation of both inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3 will bind to the endoplasmic reticulum (ER), causing ER Ca2+ efflux. DAG will activate protein kinase C (PKC). The activation of Gs-protein-coupled receptors (Blue) will activate transmembrane adenylyl cyclase (tmAC), leading to an increase in intracellular cyclic adenosine monophosphate (cAMP) production, which will activate protein kinase A (PKA). The activation of pituitary adenylate cyclase-activating polypeptide type 1 (PAC1) and endothelin (ET) receptors (Pink) could activate both Gq and Gs mechanisms. Stimulation of Gi-protein-coupled receptors (Gray) predominantly by dopamine will inhibit tmAC activity, which in turn decreases cAMP. The overall balance between the concentration of external ligands and the extent of activation of each of the Gs, Gi and Gq pathways is capable of acutely fine-tuning the type I cell hypoxic sensitivity. Many of these receptors and signaling pathways are also involved in physiological and pathological carotid body adaptation. The dashed circle identifies the position of the CB and type I cells at the carotid bifurcation. Dashed lines denote receptor activation linking to specific downstream enzymes. Filled lines denote further downstream or upstream signaling cascades.
4. Other G-Protein Signaling Mechanisms
In addition to being a principal excitatory neurotransmitter, it has been proposed that ATP has an autocrine and paracrine function in the CB to provide a negative feedback mechanism and amplify the effects of adenosine, respectively [130][131]. ATP receptors that are also GPCRs include the P2Y1- and P2Y2-receptor subtypes which are coupled to the Gαq protein and have been identified in the CB. P2Y2-receptors are localized to the glia-like sustentacular type II cells and administration of ATP leads to an increase in [Ca2+]i and the opening of pannexin-1 channels, which allow the release of ATP [130][132]. This ATP-induced ATP release allows for further breakdown of ATP via CD73 to form adenosine, which can then act on A2A- and A2B-receptors, enhancing adenosine chemoexcitation in the CB, suggesting cross-communication between type I and type II cells [1]. However, ATP can also act on P2Y1-receptors located on the type I cell, providing a negative feedback mechanism at high extracellular concentrations. Studies have shown that high extracellular levels of ATP had no effect on the resting [Ca2+]i but were able to restrict the increase in [Ca2+]i during hypoxia [131]. Although the exact mechanism is unknown, activation of the P2Y1-receptor has been suggested to close background ion channels other than the common TASK-like K+ or Na+ channels, resulting in type I cell hyperpolarization [131].
Similarly, as well as being a key excitatory neurotransmitter, acetylcholine (ACh) also has a modulatory function acting through Gαq-coupled muscarinic receptors [133][134]. Interestingly, stimulation of type I cells by methylcholine can be inhibited by histamine, H3-receptor (Gαi) agonists and SQ22536, suggestive of interaction between Gαi and Gαq pathways [135][136]. The precise point(s) at which multiple different G-protein pathways converge remains to be revealed. Exploring the cross-talk between multiple different GPCRs and downstream signaling pathways could be an interesting area for investigation in CB-mediated cardiovascular disease [73].
This entry is adapted from the peer-reviewed paper 10.3390/ijms21176012
References
- Piskuric, N.A.; Nurse, C.A. Expanding role of ATP as a versatile messenger at carotid and aortic body chemoreceptors. J. Physiol. 2013, 591, 415–422.
- Conde, S.V.; Monteiro, E.C. Hypoxia induces adenosine release from the rat carotid body. J. Neurochem. 2004, 89, 1148–1156.
- Conde, S.V.; Monteiro, E.C.; Rigual, R.; Obeso, A.; Gonzalez, C. Hypoxic intensity: A determinant for the contribution of ATP and adenosine to the genesis of carotid body chemosensory activity. J. Appl. Physiol. 2012, 112, 2002–2010.
- Kobayashi, S.; Conforti, L.; Millhorn, D.E. Gene expression and function of adenosine A(2A) receptor in the rat carotid body. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L273–L282.
- Livermore, S.; Nurse, C.A. Enhanced adenosine A2b receptor signaling facilitates stimulus-induced catecholamine secretion in chronically hypoxic carotid body type I cells. Am. J. Physiol. Cell Physiol. 2013, 305, C739–C750.
- Conde, S.V.; Obeso, A.; Vicario, I.; Rigual, R.; Rocher, A.; Gonzalez, C. Caffeine inhibition of rat carotid body chemoreceptors is mediated by A2A and A2B adenosine receptors. J. Neurochem. 2006, 98, 616–628.
- Gauda, E.B.; Northington, F.J.; Linden, J.; Rosin, D.L. Differential expression of a(2a), A(1)-adenosine and D(2)-dopamine receptor genes in rat peripheral arterial chemoreceptors during postnatal development. Brain Res. 2000, 872, 1–10.
- Monteiro, E.C.; Ribeiro, J.A. Ventilatory effects of adenosine mediated by carotid body chemoreceptors in the rat. Naunyn. Schmiedebergs Arch Pharmacol. 1987, 335, 143–148.
- Tubek, S.; Niewinski, P.; Reczuch, K.; Janczak, D.; Rucinski, A.; Paleczny, B.; Engelman, Z.J.; Banasiak, W.; Paton, J.F.; Ponikowski, P. Effects of selective carotid body stimulation with adenosine in conscious humans. J. Physiol. 2016, 594, 6225–6240.
- McQueen, D.S.; Ribeiro, J.A. Effects of beta-endorphin, vasoactive intestinal polypeptide and cholecystokinin octapeptide on cat carotid chemoreceptor activity. Q J. Exp. Physiol. 1981, 66, 273–284.
- Vandier, C.; Conway, A.F.; Landauer, R.C.; Kumar, P. Presynaptic action of adenosine on a 4-aminopyridine-sensitive current in the rat carotid body. J. Physiol. 1999, 515, 419–429.
- Xu, F.; Xu, J.; Tse, F.W.; Tse, A. Adenosine stimulates depolarization and rise in cytoplasmic [Ca2+] in type I cells of rat carotid bodies. Am. J. Physiol. Cell Physiol. 2006, 290, C1592–C1598.
- Holmes, A.P.; Nunes, A.R.; Cann, M.J.; Kumar, P. Ecto-5'-Nucleotidase, Adenosine and Transmembrane Adenylyl Cyclase Signalling Regulate Basal Carotid Body Chemoafferent Outflow and Establish the Sensitivity to Hypercapnia. In Arterial Chemoreceptors in Physiology and Pathophysiology; Peers, C., Kumar, P., Wyatt, C.N., Gauda, E., Nurse, C.A., Prabhakar, N., Eds.; Springer-Verlag Berlin: Berlin, Germany, 2015; Volume 860, pp. 279–289.
- Sacramento, J.F.; Gonzalez, C.; Gonzalez-Martin, M.C.; Conde, S.V. Adenosine Receptor Blockade by Caffeine Inhibits Carotid Sinus Nerve Chemosensory Activity in Chronic Intermittent Hypoxic Animals. Adv. Exp. Med.Biol. 2015, 860, 133–137.
- Conde, S.V.; Monteiro, E.C. Profiles for ATP and adenosine release at the carotid body in response to O2 concentrations. Adv. Exp. Med. Biol. 2006, 580, 179–184; discussion 351–359.
- McQueen, D.S.; Ribeiro, J.A. Pharmacological characterization of the receptor involved in chemoexcitation induced by adenosine. Br. J. Pharmacol. 1986, 88, 615–620.
- Holmes, A.P.; Ray, C.J.; Pearson, S.A.; Coney, A.M.; Kumar, P. Ecto-5'-nucleotidase (CD73) regulates peripheral chemoreceptor activity and cardiorespiratory responses to hypoxia. J. Physiol. London 2018, 596, 3137–3148.
- Sacramento, J.F.; Melo, B.F.; Conde, S.V. Adenosine Mediates Hypercapnic Response in the Rat Carotid Body via A2A and A2B Receptors. Adv. Exp. Med. Biol. 2018, 1071, 89–93.
- Chen, J.; Dinger, B.; Fidone, S.J. cAMP production in rabbit carotid body: Role of adenosine. J. Appl. Physiol. 1997, 82, 1771–1775.
- Conde, S.V.; Gonzalez, C.; Batuca, J.R.; Monteiro, E.C.; Obeso, A. An antagonistic interaction between A2B adenosine and D2 dopamine receptors modulates the function of rat carotid body chemoreceptor cells. J. Neurochem. 2008, 107, 1369–1381.
- Rocher, A.; Caceres, A.I.; Almaraz, L.; Gonzalez, C. EPAC signalling pathways are involved in low PO2 chemoreception in carotid body chemoreceptor cells. J. Physiol. 2009, 587, 4015–4027.
- Zhang, M.; Vollmer, C.; Nurse, C.A. Adenosine and dopamine oppositely modulate a hyperpolarization-activated current I(h) in chemosensory neurons of the rat carotid body in co-culture. J. Physiol. 2018, 596, 3101–3117.
- Zhang, M.; Nurse, C.A. CO2/pH chemosensory signaling in co-cultures of rat carotid body receptors and petrosal neurons: Role of ATP and ACh. J. Neurophysiol. 2004, 92, 3433–3445.
- Murali, S.; Nurse, C.A. Purinergic signalling mediates bidirectional crosstalk between chemoreceptor typeI and glial-like typeII cells of the rat carotid body. J. Physiol. London 2016, 594, 391–406.
- Bianchi, V.; Spychala, J. Mammalian 5'-nucleotidases. J. Biol. Chem. 2003, 278, 46195–46198.
- Salman, S.; Vollmer, C.; McClelland, G.B.; Nurse, C.A. Characterization of ectonucleotidase expression in the rat carotid body: Regulation by chronic hypoxia. Am. J. Physiol. Cell Physiol. 2017, 313, C274–C284.
- Li, J.; Wang, L.; Chen, X.; Li, L.; Li, Y.; Ping, Y.; Huang, L.; Yue, D.; Zhang, Z.; Wang, F.; et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 2017, 6, e1320011.
- Salman, S.; Nurse, C.A. Molecular Characterization of Equilibrative Nucleoside Transporters in the Rat Carotid Body and Their Regulation by Chronic Hypoxia. Adv. Exp. Med. Biol. 2018, 1071, 43–50.
- Sacramento, J.F.; Olea, E.; Ribeiro, M.J.; Prieto-Lloret, J.; Melo, B.F.; Gonzalez, C.; Martins, F.O.; Monteiro, E.C.; Conde, S.V. Contribution of adenosine and ATP to the carotid body chemosensory activity in ageing. J. Physiol. 2019, 597, 4991–5008.
- de Lucia, C.; Eguchi, A.; Koch, W.J. New Insights in Cardiac β-Adrenergic Signaling During Heart Failure and Aging. Front Pharm. 2018, 9, 904–904.
- Joels, N.; White, H. The contribution of the arterial chemoreceptors to the stimulation of respiration by adrenaline and noradrenaline in the cat. J. Physiol. 1968, 197, 1–23.
- Thompson, E.L.; Ray, C.J.; Holmes, A.P.; Pye, R.L.; Wyatt, C.N.; Coney, A.M.; Kumar, P. Adrenaline release evokes hyperpnoea and an increase in ventilatory CO2 sensitivity during hypoglycaemia: A role for the carotid body. J. Physiol. 2016, 594, 4439–4452.
- Folgering, H.; Ponte, J.; Sadig, T. Adrenergic mechanisms and chemoreception in the carotid body of the cat and rabbit. J. Physiol. 1982, 325, 1–21.
- Jones, J.F.X. Retrospective view of the carotid body research of Ronan G. O′Regan. Exp. Physiol. 2004, 89, 39–43.
- Hauton, D.; Holmes, A.; Ziff, O.; Kumar, P. The impact of acute and chronic catecholamines on respiratory responses to hypoxic stress in the rat. Pflugers Arch. 2013, 465, 209–219.
- Ward, D.S.; Voter, W.A.; Karan, S. The effects of hypo- and hyperglycaemia on the hypoxic ventilatory response in humans. J. Physiol. 2007, 582, 859–869.
- Koyama, Y.; Coker, R.H.; Stone, E.E.; Lacy, D.B.; Jabbour, K.; Williams, P.E.; Wasserman, D.H. Evidence that carotid bodies play an important role in glucoregulation in vivo. Diabetes 2000, 49, 1434–1442.
- Bin-Jaliah, I.; Maskell, P.D.; Kumar, P. Indirect sensing of insulin-induced hypoglycaemia by the carotid body in the rat. J. Physiol. 2004, 556, 255–266.
- Wehrwein, E.A.; Basu, R.; Basu, A.; Curry, T.B.; Rizza, R.A.; Joyner, M.J. Hyperoxia blunts counterregulation during hypoglycaemia in humans: Possible role for the carotid bodies? J. Physiol. 2010, 588, 4593–4601.
- Wehrwein, E.A.; Limberg, J.K.; Taylor, J.L.; Dube, S.; Basu, A.; Basu, R.; Rizza, R.A.; Curry, T.B.; Joyner, M.J. Effect of bilateral carotid body resection on the counterregulatory response to hypoglycaemia in humans. Exp. Physiol. 2015, 100, 69–78.
- Bin-Jaliah, I.; Maskell, P.D.; Kumar, P. Carbon dioxide sensitivity during hypoglycaemia-induced, elevated metabolism in the anaesthetized rat. J. Physiol. 2005, 563, 883–893.
- Holmes, A.P.; Ray, C.J.; Thompson, E.L.; Alshehri, Z.; Coney, A.M.; Kumar, P. Adrenaline activation of the carotid body: Key to CO2 and pH homeostasis in hypoglycaemia and potential pathological implications in cardiovascular disease. Respir. Physiol. Neurobiol. 2019, 265, 92–99.
- Chang, A.J.; Ortega, F.E.; Riegler, J.; Adison, D.V.M.; Krasnow, M.A. Oxygen regulation of breathing through an olfactory receptor activated by lactate. Nature 2015, 527, 240.
- Peng, Y.-J.; Gridina, A.; Wang, B.; Nanduri, J.; Fox, A.P.; Prabhakar, N.R. Olfactory receptor 78 participates in carotid body response to a wide range of low O2 levels but not severe hypoxia. J. Neurophysiol. 2020, 123, 1886–1895.
- Torres-Torrelo, H.; Ortega-Sáenz, P.; Macías, D.; Omura, M.; Zhou, T.; Matsunami, H.; Johnson, R.S.; Mombaerts, P.; López-Barneo, J. The role of Olfr78 in the breathing circuit of mice. Nature 2018, 561, E33–E40.
- Holmes, A.P.; Turner, P.J.; Carter, P.; Leadbeater, W.; Ray, C.J.; Hauton, D.; Buckler, K.J.; Kumar, P. Glycogen metabolism protects against metabolic insult to preserve carotid body function during glucose deprivation. J. Physiol. 2014, 592, 4493–4506.
- Brown, A.M.; Evans, R.D.; Black, J.; Ransom, B.R. Schwann cell glycogen selectively supports myelinated axon function. Ann. Neurol. 2012, 72, 406–418.
- Tekkok, S.B.; Brown, A.M.; Westenbroek, R.; Pellerin, L.; Ransom, B.R. Transfer of glycogen-derived lactate from astrocytes to axons via specific monocarboxylate transporters supports mouse optic nerve activity. J. Neurosci. Res. 2005, 81, 644–652.
- Zapata, P.; Hess, A.; Bliss, E.L.; Eyzaguirre, C. Chemical, electron microscopic and physiological observations on the role of catecholamines in the carotid body. Brain Res. 1969, 14, 473–496.
- Nurse, C.A.; Fearon, I.M. Carotid body chemoreceptors in dissociated cell culture. Microsc. Res. Tech. 2002, 59, 249–255.
- Chiocchio, S.R.; Biscardi, A.M.; Tramezzani, J.H. Catecholamines in the carotid body of the cat. Nature 1966, 212, 834–835.
- Yokoyama, T.; Yamamoto, Y.; Hirakawa, M.; Kato, K.; Saino, T. Vesicular nucleotide transporter-immunoreactive type I cells associated with P2X3-immunoreactive nerve endings in the rat carotid body. J. Comp. Neurol. 2020, 528, 1486–1501.
- Kato, K.; Fushuku, S.; Yamamoto, Y. Age-related changes in immunoreactivity for dopamine beta-hydroxylase in carotid body glomus cells in spontaneously hypertensive rats. Auton. Neurosci-Basic Clin. 2017, 205, 50–56.
- Ortega-Saenz, P.; Macias, D.; Levitsky, K.L.; Rodriguez-Gomez, J.A.; Gonzalez-Rodriguez, P.; Bonilla-Henao, V.; Arias-Mayenco, I.; Lopez-Barneo, J. Selective accumulation of biotin in arterial chemoreceptors: Requirement for carotid body exocytotic dopamine secretion. J. Physiol. London 2016, 594, 7229–7248.
- Kato, K.; Yokoyama, T.; Kusakabe, T.; Hata, K.; Fushuku, S.; Nakamuta, N.; Yamamoto, Y. Differences in the expression of catecholamine-synthesizing enzymes between vesicular monoamine transporter 1-and 2-immunoreactive glomus cells in the rat carotid body. Acta Histochem. 2020, 122, 9.
- Fidone, S.; Gonzalez, C.; Yoshizaki, K. Effects of low oxygen on the release of dopamine from the rabbit carotid body in vitro. J. Physiol. 1982, 333, 93–110.
- Urena, J.; Fernandez-Chacon, R.; Benot, A.R.; Alvarez de Toledo, G.A.; Lopez-Barneo, J. Hypoxia induces voltage-dependent Ca2+ entry and quantal dopamine secretion in carotid body glomus cells. Proc. Natl. Acad. Sci. USA 1994, 91, 10208–10211.
- Wakai, J.; Takayama, A.; Yokoyama, T.; Nakamuta, N.; Kusakabe, T.; Yamamoto, Y. Immunohistochemical localization of dopamine D2 receptor in the rat carotid body. Acta Histochem. 2015, 117, 784–789.
- Welsh, M.J.; Heistad, D.D.; Abboud, F.M. Depression of ventilation by dopamine in man. Evidence for an effect on the chemoreceptor reflex. J. Clin. Investig. 1978, 61, 708–713.
- Zapata, P. Effects of dopamine on carotid chemo- and baroreceptors in vitro. J. Physiol. 1975, 244, 235–251.
- Docherty, R.J.; McQueen, D.S. Inhibitory action of dopamine on cat carotid chemoreceptors. J. Physiol. 1978, 279, 425–436.
- Carroll, J.L.; Boyle, K.M.; Wasicko, M.J.; Sterni, L.M. Dopamine D2 receptor modulation of carotid body type 1 cell intracellular calcium in developing rats. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L910–L916.
- Fagerlund, M.J.; Kahlin, J.; Ebberyd, A.; Schulte, G.; Mkrtchian, S.; Eriksson, L.I. The Human Carotid Body Expression of Oxygen Sensing and Signaling Genes of Relevance for Anesthesia. Anesthesiology 2010, 113, 1270–1279.
- Johnson, B.D.; Peinado, A.B.; Ranadive, S.M.; Curry, T.B.; Joyner, M.J. Effects of intravenous low-dose dopamine infusion on glucose regulation during prolonged aerobic exercise. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R49–R57.
- Limberg, J.K.; Johnson, B.D.; Mozer, M.T.; Holbein, W.W.; Curry, T.B.; Prabhakar, N.R.; Joyner, M.J. Role of the carotid chemoreceptors in insulin-mediated sympathoexcitation in humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R173–R181.
- Phillips, D.B.; Steinback, C.D.; Collins, S.E.; Fuhr, D.P.; Bryan, T.L.; Wong, E.Y.L.; Tedjasaputra, V.; Bhutani, M.; Stickland, M.K. The carotid chemoreceptor contributes to the elevated arterial stiffness and vasoconstrictor outflow in chronic obstructive pulmonary disease. J. Physiol. London 2018, 596, 3233–3244.
- Bain, A.R.; Dujic, Z.; Hoiland, R.L.; Barak, O.F.; Madden, D.; Drvis, I.; Stembridge, M.; MacLeod, D.B.; MacLeod, D.M.; Ainslie, P.N. Peripheral chemoreflex inhibition with low-dose dopamine: New insight into mechanisms of extreme apnea. Am. J. Physiol.-Regul. Integr. Comp. Physiol.2015, 309, R1162–R1171.
- Limberg, J.K.; Johnson, B.D.; Holbein, W.W.; Ranadive, S.M.; Mozer, M.T.; Joyner, M.J. Interindividual variability in the dose-specific effect of dopamine on carotid chemoreceptor sensitivity to hypoxia. J. Appl. Physiol. 2016, 120, 138–147.
- Prieto-Lloret, J.; Donnelly, D.F.; Rico, A.J.; Moratalla, R.; Gonzalez, C.; Rigual, R.J. Hypoxia transduction by carotid body chemoreceptors in mice lacking dopamine D(2) receptors. J. Appl. Physiol. 2007, 103, 1269–1275.
- Bialkowska, M.; Zajac, D.; Mazzatenta, A.; Di Giulio, C.; Pokorski, M. Inhibition of Peripheral Dopamine Metabolism and the Ventilatory Response to Hypoxia in the Rat. In Neurotransmitter Interactions and Cognitive Function; Pokorski, M., Ed.; Springer-Verlag Berlin: Berlin, Germany, 2015; Volume 837, pp. 9–17.
- Huey, K.A.; Powell, F.L. Time-dependent changes in dopamine D(2)-receptor mRNA in the arterial chemoreflex pathway with chronic hypoxia. Brain Res. Mol. Brain Res. 2000, 75, 264–270.
- Wakai, J.; Kizaki, K.; Yamaguchi-Yamada, M.; Yamamoto, Y. Differences in tyrosine hydroxylase expression after short-term hypoxia, hypercapnia or hypercapnic hypoxia in rat carotid body. Respir. Physiol. Neurobiol. 2010, 173, 95–100.
- Porzionato, A.; Stocco, E.; Guidolin, D.; Agnati, L.; Macchi, V.; De Caro, R. Receptor-Receptor Interactions of G Protein-Coupled Receptors in the Carotid Body: A Working Hypothesis. Front. Physiol. 2018, 9, 697–697.
- Olea, E.; Docio, I.; Quintero, M.; Rocher, A.; Obeso, A.; Rigual, R.; Gomez-Niño, A. Peripheral Dopamine 2-Receptor Antagonist Reverses Hypertension in a Chronic Intermittent Hypoxia Rat Model. Int. J. Mol. Sci. 2020, 21, 4893.
- Gomeznino, A.; Dinger, B.; Gonzalez, C.; Fidone, S.J. Differential Stimulus Coupling To Dopamine And Norepinephrine Stores In Rabbit Carotid-Body Type-I Cells. Brain Res. 1990, 525, 160–164.
- Schamel, A.; Verna, A. Norepinephrine-containing glomus cells in the rabbit carotid-body.2. immunocytochemical evidence of dopamine-beta-hydroxylase and norepinephrine. J. Neurocytol. 1992, 21, 353–362.
- Bisgard, G.E.; Mitchell, R.A.; Herbert, D.A. Effects of dopamine, norepinephrine and 5-hydroxytryptamine on the carotid body of the dog. Respir. Physiol. 1979, 37, 61–80.
- Kou, Y.R.; Ernsberger, P.; Cragg, P.A.; Cherniack, N.S.; Prabhakar, N.R. Role of alpha 2-adrenergic receptors in the carotid body response to isocapnic hypoxia. Respir. Physiol. 1991, 83, 353–364.
- Pizarro, J.; Warner, M.M.; Ryan, M.; Mitchell, G.S.; Bisgard, G.E. Intracarotid norepinephrine infusions inhibit ventilation in goats. Respir. Physiol. 1992, 90, 299–310.
- Prabhakar, N.R.; Kou, Y.R. Inhibitory sympathetic action on the carotid body responses to sustained hypoxia. Respir. Physiol. 1994, 95, 67–79.
- Overholt, J.L.; Prabhakar, N.R. Norepinephrine inhibits a toxin resistant Ca2+ current in carotid body glomus cells: Evidence for a direct G protein mechanism. J. Neurophysiol. 1999, 81, 225–233.
- Almaraz, L.; Perez-Garcia, M.T.; Gomez-Nino, A.; Gonzalez, C. Mechanisms of alpha2-adrenoceptor-mediated inhibition in rabbit carotid body. Am. J. Physiol. 1997, 272, C628–C637
- Allen, A.M. Angiotensin AT(1) receptor-mediated excitation of rat carotid body chemoreceptor afferent activity. J. Physiol. London 1998, 510, 773–781.
- Peng, Y.J.; Raghuraman, G.; Khan, S.A.; Kumar, G.K.; Prabhakar, N.R. Angiotensin II evokes sensory long-term facilitation of the carotid body via NADPH oxidase. J. Appl. Physiol. 2011, 111, 964–970.
- Fung, M.L.; Lam, S.Y.; Chen, Y.; Dong, X.; Leung, P.S. Functional expression of angiotensin II receptors in type-I cells of the rat carotid body. Pflugers Arch. 2001, 441, 474–480.
- Atanasova, D.Y.; Dandov, A.D.; Dimitrov, N.D.; Lazarov, N.E. Immunohistochemical localization of angiotensin AT(1) receptors in the rat carotid body. Acta Histochem. 2018, 120, 154–158.
- Leung, P.S.; Fung, M.L.; Tam, M.S. Renin-angiotensin system in the carotid body. Int. J. Biochem. Cell Biol. 2003, 35, 847–854.
- Wang, J.J.; Hogan, J.O.; Kim, D. Voltage- and receptor-mediated activation of a non-selective cation channel in rat carotid body glomus cells. Respir. Physiol. Neuro. 2017, 237, 13–21.
- Kim, S.J.; Fong, A.Y.; Pilowsky, P.M.; Abbott, S.B.G. Sympathoexcitation following intermittent hypoxia in rat is mediated by circulating angiotensin II acting at the carotid body and subfornical organ. J. Physiol. London 2018, 596, 3217–3232.
- Roy, A.; Farnham, M.M.J.; Derakhshan, F.; Pilowsky, P.M.; Wilson, R.J.A. Acute intermittent hypoxia with concurrent hypercapnia evokes P2X and TRPV1 receptor-dependent sensory long-term facilitation in naive carotid bodies. J. Physiol. 2018, 596, 3149–3169.
- Brown, C.V.; Boulet, L.M.; Vermeulen, T.D.; Sands, S.A.; Wilson, R.J.A.; Ayas, N.T.; Floras, J.S.; Foster, G.E. Angiotensin II-Type I Receptor Antagonism Does Not Influence the Chemoreceptor Reflex or Hypoxia-Induced Central Sleep Apnea in Men. Front. Neurosci. 2020, 14, 13.
- Solaiman, A.Z.; Feehan, R.P.; Chabitnoy, A.M.; Leuenberger, U.A.; Monahan, K.D. Ventilatory responses to chemoreflex stimulation are not enhanced by angiotensin II in healthy humans. Auton. Neurosci-Basic Clin. 2014, 183, 72–79.
- Leung, P.S.; Lam, S.Y.; Fung, M.L. Chronic hypoxia upregulates the expression and function of AT(1) receptor in rat carotid body. J. Endocrinol. 2000, 167, 517–524.
- Lam, S.Y.; Fung, M.L.; Leung, P.S. Regulation of the angiotensin-converting enzyme activity by a time-course hypoxia in the carotid body. J. Appl. Physiol. 2004, 96, 809–813.
- Lam, S.Y.; Liu, Y.; Ng, K.M.; Liong, E.C.; Tipoe, G.L.; Leung, P.S.; Fung, M.L. Upregulation of a local renin-angiotensin system in the rat carotid body during chronic intermittent hypoxia. Exp. Physiol. 2014, 99, 220–231.
- Marcus, N.J.; Li, Y.L.; Bird, C.E.; Schultz, H.D.; Morgan, B.J. Chronic intermittent hypoxia augments chemoreflex control of sympathetic activity: Role of the angiotensin II type 1 receptor. Respir. Physiol. Neurobiol. 2010, 171, 36–45.
- Morgan, B.J.; Teodorescu, M.; Pegelow, D.F.; Jackson, E.R.; Schneider, D.L.; Plante, D.T.; Gapinski, J.P.; Hetzel, S.J.; Dopp, J.M. Effects of losartan and allopurinol on cardiorespiratory regulation in obstructive sleep apnoea. Exp. Physiol. 2018, 103, 941–955.
- Nanduri, J.; Peng, Y.J.; Wang, N.; Khan, S.A.; Semenza, G.L.; Kumar, G.K.; Prabhakar, N.R. Epigenetic regulation of redox state mediates persistent cardiorespiratory abnormalities after long-term intermittent hypoxia. J. Physiol. London 2017, 595, 63–77.
- Li, Y.L.; Xia, X.H.; Zheng, H.; Gao, L.; Li, Y.F.; Liu, D.; Patel, K.P.; Wang, W.; Schultz, H.D. Angiotensin II enhances carotid body chemoreflex control of sympathetic outflow in chronic heart failure rabbits. Cardiovasc Res. 2006, 71, 129–138.
- Li, Y.L.; Schultz, H.D. Enhanced sensitivity of Kv channels to hypoxia in the rabbit carotid body in heart failure: Role of angiotensin II. J. Physiol. 2006, 575, 215–227
- Li, Y.L.; Gao, L.; Zucker, I.H.; Schultz, H.D. NADPH oxidase-derived superoxide anion mediates angiotensin II-enhanced carotid body chemoreceptor sensitivity in heart failure rabbits. Cardiovasc Res. 2007, 75, 546–554.
- Oomori, Y.; Nakaya, K.; Tanaka, H.; Iuchi, H.; Ishikawa, K.; Satoh, Y.; Ono, K. Immunohistochemical and histochemical evidence for the presence of noradrenaline, serotonin and gamma-aminobutyric acid in chief cells of the mouse carotid body. Cell Tissue Res. 1994, 278, 249–254.
- Yokoyama, T.; Misuzu, Y.Y.; Yamamoto, Y. Immunohistochemical localization of tryptophan hydroxylase and serotonin transporter in the carotid body of the rat. Histochem. Cell Biol. 2013, 140, 147–155.
- Zhang, M.; Nurse, C.A. Does endogenous 5-HT mediate spontaneous rhythmic activity in chemoreceptor clusters of rat carotid body? Brain Res. 2000, 872, 199–203.
- Zhang, M.; Fearon, I.M.; Zhong, H.; Nurse, C.A. Presynaptic modulation of rat arterial chemoreceptor function by 5-HT: Role of K+ channel inhibition via protein kinase C. J. Physiol. 2003, 551, 825–842.
- Murali, S.; Zhang, M.; Nurse, C.A. Evidence that 5-HT stimulates intracellular Ca2+ signalling and activates pannexin-1 currents in type II cells of the rat carotid body. J. Physiol. 2017, 595, 4261–4277.
- Yokoyama, T.; Nakamuta, N.; Kusakabe, T.; Yamamoto, Y. Serotonin-mediated modulation of hypoxia-induced intracellular calcium responses in glomus cells isolated from rat carotid body. Neurosci. Lett. 2015, 597, 149–153.
- Jacono, F.J.; Peng, Y.J.; Kumar, G.K.; Prabhakar, N.R. Modulation of the hypoxic sensory response of the carotid body by 5-hydroxytryptamine: Role of the 5-HT2 receptor. Respir. Physiol. Neurobiol. 2005, 145, 135–142.
- Peng, Y.J.; Yuan, G.; Jacono, F.J.; Kumar, G.K.; Prabhakar, N.R. 5-HT evokes sensory long-term facilitation of rodent carotid body via activation of NADPH oxidase. J. Physiol. 2006, 576, 289–295.
- Peng, Y.J.; Nanduri, J.; Yuan, G.; Wang, N.; Deneris, E.; Pendyala, S.; Natarajan, V.; Kumar, G.K.; Prabhakar, N.R. NADPH oxidase is required for the sensory plasticity of the carotid body by chronic intermittent hypoxia. J. Neurosci. 2009, 29, 4903–4910.
- Koves, K.; Szabo, E.; Kantor, O.; Heinzlmann, A.; Szabo, F.; Csaki, A. Current State of Understanding of the Role of PACAP in the Hypothalamo-Hypophyseal Gonadotropin Functions of Mammals. Front. Endocrinol. 2020, 11, 21.
- Rita, B.; Laszlo, M.; Andrea, T.; Terez, B.; Csaba, B.; Katalin, C.; Eszter, B.; Peter, K.; Alexandra, V.; Gabriella, H.; et al. Presence of pituitary adenylate cyclase activating polypeptide-38 in human plasma and milk. Eur. J. Endocrinol. 2009, 160, 561–565.
- Xu, F.; Tse, F.W.; Tse, A. Pituitary adenylate cyclase-activating polypeptide (PACAP) stimulates the oxygen sensing type I (glomus) cells of rat carotid bodies via reduction of a background TASK-like K+ current. J. Neurochem. 2007, 101, 1284–1293.
- Roy, A.; Derakhshan, F.; Wilson, R.J.A. Stress peptide PACAP engages multiple signaling pathways within the carotid body to initiate excitatory responses in respiratory and sympathetic chemosensory afferents. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2013, 304, R1070–R1084.
- Cummings, K.J.; Klotz, C.; Liu, W.Q.; Weese-Mayer, D.E.; Marazita, M.L.; Cooper, M.E.; Berry-Kravis, E.M.; Tobias, R.; Goldie, C.; Bech-Hansen, N.T.; et al. Sudden infant death syndrome (SIDS) in African Americans: Polymorphisms in the gene encoding the stress peptide pituitary adenylate cyclase-activating polypeptide (PACAP). Acta Paediatr. 2009, 98, 482–489.
- Cummings, K.J.; Pendlebury, J.D.; Jirik, F.R.; Sherwood, N.M.; Wilson, R.J.A. A SIDS-like phenotype is associated with reduced respiratory chemoresponses in PACAP deficient neonatal mice. In Post-Genomic Perspectives in Modeling and Control of Breathing; Champagnat, J., DenavitSaubie, M., Fortin, G., ThobyBrisson, M., Eds.; Kluwer Academic/Plenum Publ: New York, NY, USA, 2004; Volume 551, pp. 77–83.
- Barrett, K.T.; Hasan, S.U.; Scantlebury, M.H.; Wilson, R.J.A. Impaired neonatal cardiorespiratory responses to hypoxia in mice lacking PAC1 or VPAC2 receptors. Am. J. Physiol.-Regul. Integr. Comp. Physiol.2019, 316, R594–R606.
- Ferguson, E.V.; Roy, A.; Ciechanski, P.; Wilson, R.J.A. Stress peptide PACAP stimulates and stabilizes neonatal breathing through distinct mechanisms. Respir. Physiol. Neuro. 2013, 187, 217–223.
- Rey, S.; Del Rio, R.; Iturriaga, R. Role of endothelin-1 on the enhanced carotid body activity induced by chronic intermittent hypoxia. Adv. Exp. Med. Biol. 2006, 580, 345–350; discussion 351–359.
- Chen, J.; He, L.; Dinger, B.; Fidone, S. Cellular mechanisms involved in rabbit carotid body excitation elicited by endothelin peptides. Respir. Physiol. 2000, 121, 13–23.
- Chen, Y.; Tipoe, G.L.; Liong, E.; Leung, S.; Lam, S.Y.; Iwase, R.; Tjong, Y.W.; Fung, M.L. Chronic hypoxia enhances endothelin-1-induced intracellular calcium elevation in rat carotid body chemoreceptors and up-regulates ETA receptor expression. Pflugers Arch. 2002, 443, 565–573.
- Rey, S.; Del Rio, R.; Iturriaga, R. Role of endothelin-1 on the enhanced carotid body activity induced by chronic intermittent hypoxia. Adv. Exp. Med. Biol. 2006, 580, 345–350; discussion 351–359
- Pawar, A.; Nanduri, J.; Yuan, G.; Khan, S.A.; Wang, N.; Kumar, G.K.; Prabhakar, N.R. Reactive oxygen species-dependent endothelin signaling is required for augmented hypoxic sensory response of the neonatal carotid body by intermittent hypoxia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R735–R742.
- Mosqueira, M.; Iturriaga, R. Chronic hypoxia changes gene expression profile of primary rat carotid body cells: Consequences on the expression of NOS isoforms and ET-1 receptors. Physiol. Genomics 2019, 51, 109–124.
- Chen, J.; He, L.; Dinger, B.; Stensaas, L.; Fidone, S. Role of endothelin and endothelin A-type receptor in adaptation of the carotid body to chronic hypoxia. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 282, L1314–L1323
- Holmes, A.P.; Turner, P.J.; Buckler, K.J.; Kumar, P. Moderate inhibition of mitochondrial function augments carotid body hypoxic sensitivity. Pflugers Arch. 2016, 468, 143–155.
- Peng, Y.J.; Nanduri, J.; Raghuraman, G.; Wang, N.; Kumar, G.K.; Prabhakar, N.R. Role of oxidative stress-induced endothelin-converting enzyme activity in the alteration of carotid body function by chronic intermittent hypoxia. Exp. Physiol. 2013, 98, 1620–1630.
- Rey, S.; Del Rio, R.; Iturriaga, R. Contribution of endothelin-1 to the enhanced carotid body chemosensory responses induced by chronic intermittent hypoxia. Brain Res. 2006, 1086, 152–159.
- Li, J.; Yang, S.; Yu, F.; Ji, E.; Woodrow Weiss, J. Endothelin-1 enhanced carotid body chemosensory activity in chronic intermittent hypoxia through PLC, PKC and p38MAPK signaling pathways. Neuropeptides 2019, 74, 44–51.
- Xu, J.; Tse, F.W.; Tse, A. ATP triggers intracellular Ca2+ release in type II cells of the rat carotid body. J. Physiol. 2003, 549, 739–747.
- Xu, J.; Xu, F.; Tse, F.W.; Tse, A. ATP inhibits the hypoxia response in type I cells of rat carotid bodies. J. Neurochem. 2005, 92, 1419–1430.
- Zhang, M.; Piskuric, N.A.; Vollmer, C.; Nurse, C.A. P2Y2 receptor activation opens pannexin-1 channels in rat carotid body type II cells: Potential role in amplifying the neurotransmitter ATP. J. Physiol. London 2012, 590, 4335–4350
- Bairam, A.; Joseph, V.; Lajeunesse, Y.; Kinkead, R. Developmental pattern of M1 and M2 muscarinic gene expression and receptor levels in cat carotid body, petrosal and superior cervical ganglion. Neuroscience 2006, 139, 711–721.
- Shirahata, M.; Hirasawa, S.; Okumura, M.; Mendoza, J.A.; Okumura, A.; Balbir, A.; Fitzgerald, R.S. Identification of M1 and M2 muscarinic acetylcholine receptors in the cat carotid body chemosensory system. Neuroscience 2004, 128, 635–644.
- Thompson, C.M.; Wyatt, C.N. Inhibition of adenylate cyclase attenuates muscarinic Ca(2)(+) signaling by a PKA-independent mechanism in rat carotid body Type I cells. Respir. Physiol. Neurobiol. 2011, 175, 90–96.
- Thompson, C.M.; Troche, K.; Jordan, H.L.; Barr, B.L.; Wyatt, C.N. Evidence for functional, inhibitory, histamine H3 receptors in rat carotid body Type I cells. Neurosci. Lett. 2010, 471, 15–19.