Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Health Care Sciences & Services
Organoselenium compounds have anti-oxidative effects, several natural and synthetic organoselenium compounds and metabolites act as histone deacetylase inhibitors, which influence the acetylation status of histones and non-histone proteins, altering gene transcription.
- organoselenium compounds
1. Synthetic Organoselenium Compounds
The synthesis of organoselenium compounds was first reported by Lowig as early as 1836; however, the malodorous nature, troublesome purification, and the instability of many Se derivatives hampered early developments [55]. Research into organoselenium compounds picked up in the 1970s as they were found to be less toxic than their inorganic counterparts and were found to have several useful applications [55,56,57]. Presently, the synthesis and applications of organoselenium compounds are still the centre of intense research and may play a central role in cancer therapeutics [16].
2. Methylseleninic Acid
The oxoacid methylseleninic acid (MSA, CH3SeO2H) is considered among the simplest Se-containing compounds with chemopreventative and chemotherapeutic properties. Due to its pro-oxidant nature, MSA was shown to be effective against human pancreatic [58], lung [59], breast [60], and prostate [61,62] tumour cellular models. MSA has also shown to be effective against rodent mammary [63] and pancreatic [58] in vivo cancer models, as well as colon [64] and prostate cancer [54,58] xenograft models.
Contrary to selenoamino acids, MSA circumvents the need for β-lyase to generate methylselenol. MSA is easily reduced to methylselenol via enzymatic and nonenzymatic processes [65]. In a reaction with three molecules of thiol, MSA forms selenylsulfide, which is further reduced to methylselenol in the presence of excess thiols [66]. In cells, where glutathione is the major thiol, a methyl-selenium-glutathione intermediate is formed, which undergoes reduction by glutathione reductase to form the key intermediate methylselenol. Methylselenol can undergo demethylation to replete selenoenzymes, producing hydrogen selenide [65], or be further methylated to dimethyl selenide (Figure 4) [67]. The reduction to methylselenol generates superoxide, resulting in cellular dysfunction and death [16]. The redox modifications induced by MSA may contribute to its anti-proliferative and pro-apoptotic effects in cancer cells via caspase activation, ER stress, induction of unfolded protein response, cytochrome c, and PARP cleavage [61,62].
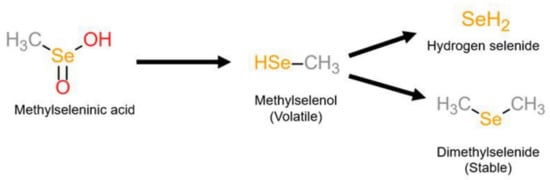
Figure 4. Metabolism of methylselenic acid (MSA). MSA is reduced to the volatile metabolite methylselenol, which is further reduced to hydrogen selenide or methylated to the stable dimethyl selenide.
In addition to the pro-oxidative properties of MSA, the inhibition of HDAC activity could be a contributing factor to MSA’s anti-carcinogenic effects. Kassam, Goenaga-Infante [67] were the first to demonstrate the HDAC inhibitory action of MSA in four diffuse large B cell lymphoma cell lines (diffuse large B-cell lymphoma (DLBCL): DoHH2, RL, SUD4, and DHL4). MSA (30 µM, 2 hr) was shown to inhibit both class I and II HDACs by 40–50% as a concentration-dependent increase in the acetylation of H3 (regulated by class I HDACs) and α -tubulin (regulated by HDAC6, a class II HDAC) occurred. HDAC activity was also measured using cell-based and cell-free assays. While the activity assays involving intact cells confirmed the concentration-dependent HDAC inhibitory action of MSA in all four cell lines, MSA did not affect HDAC activity in the cell-free assay, which used HeLa nuclear extracts. The entry further demonstrated that medium from cells exposed to MSA had a slight (21%) inhibitory effect on the HDAC activity of HeLa nuclear extracts; however, medium incubated with MSA in the absence of cells had no effect on the activity of HDACs [67]. The above data suggest that the inhibitory action of MSA is likely due to the intracellular activation of MSA to methylselenol, which is responsible for its anti-tumour activity [65]. The volatile nature of methylselenol would explain the small effect observed as it is not retained in the cell medium. Kossam, Goenaga-Infante [67] found that the intracellular Se metabolite formed after MSA exposure to the cell was dimethyl selenide. Although methylselenol was not detectable due to its high volatility, the presence of dimethyl-selenide confirmed that methylselenol was the major metabolite formed and was thus likely responsible for HDAC inhibition.
The entry further hypothesised that inhibition of HDAC activity might be responsible for HIF-1 expression and activity, providing a potential mechanism by which MSA inhibits angiogenesis. However, they did not demonstrate a direct relationship between HDAC activity and HIF-1 expression or activity. It was suggested that the concentration inhibiting HDAC activity was similar to that required for the inhibition of HIF-1α expression and VEGF secretion. Thus, HDAC inhibition may be a potential mechanism by which MSA inhibits angiogenesis in vivo, although this claim requires further investigation [67].
The modulation of HDAC activity was further investigated in human oesophageal squamous cell carcinoma cell lines (EC9760 and KYSE-150) exposed to MSA (5 µM; 24 hr) [53]. MSA treatment significantly increased H3 acetylation at lysine 9 (H3K9) and lysine 18 (H3K18); however, no detectable changes were observed at other sites on H3, and the total H3 was only slightly upregulated. H3 hyperacetylation post-MSA treatment was due to the reduced expression of HDAC 1 and 2, impaired HDAC activity, and increased expression of the HAT, general control non-repressed protein 5 (GCN5) [53]. Krüppel-like factor 4 (KLF4) participates in the transcription of various oncogenes and tumour suppressor genes and could either promote or inhibit cell growth in a tissue-dependent manner [68,69]. Overexpression of KLF4 was shown to inhibit growth and invasion of several tumour cell lines [70]; however, it is widely reported to be downregulated in ESCC. MSA treatment increased KLF4 expression via the increased acetylation of H3 at KLF4 promoters in KYSE-150 cells, contributing to MSA-mediated ESCC cell growth inhibition [71].
While the earlier studies examined specific acetylation marks, a recent study by Khalkar, Ali [72] in human chronic myeloid leukaemia K562 cells evaluated and compared genome-wide epigenetic alterations induced by MSA (5 µM, 24 hr) with those of the inorganic Se, selenite (6 µM, 24 hr). Both compounds reduced the global nuclear HDAC activity by 10%; however, these results were not significant. Western blot analysis revealed a significant increase in global H3K9ac upon MSA treatment; these results were not supported in MCF-7 breast cancer cells, which showed that MSA had no effect on H3K9ac [73]. Both studies did observe a negligible effect on H3K9ac by selenite. A chromatin immunoprecipitation assay followed by a whole genome-wide sequencing using the H3K9ac histone mark revealed that the cytotoxic effects exerted by MSA were not solely dependent on its pro-oxidant nature. MSA affected genes related to cell adhesion, glucocorticoid receptor binding, and inositol-3-phosphate synthase activity [72].
The mechanism by which MSA inhibits HDAC activity needs further investigation. Classical HDAC I and II inhibitors contain a side chain that can easily reach the catalytic pocket of HDACs to chelate Zn2+ ions found at the active site. Neither MSA nor its metabolites include these features [74]. However, MSA was shown to inhibit other enzymes, such as PKC, via redox modifications to key cysteine residues [75]. There is, therefore, the potential for Se compounds to directly alter the HDAC structure and catalytic activity; however, such a relationship needs further investigation.
3. Selenoderivatives of Suberoylanilide Hydroxamic Acid
Suberoylanilide hydroxamic acid (SAHA, C14H20N2O3) or vorinostat is a well-known HDAC inhibitor approved by the USA Food and Drug Administration for treatment against advanced cutaneous T cell lymphoma [76,77]. It has been shown to be effective against other hematological malignancies and is known to block in vitro and in vivo proliferation of cancer cells with little to no toxicity to normal cells [78,79,80,81,82]. The anti-proliferative effect of SAHA is believed to be due to its ability to inhibit HDAC activity, leading to the accumulation of acetylated proteins and histones, thus altering the transcription and activity of multiple genes related to cell cycle arrest, apoptosis, and differentiation [83,84,85]. While SAHA is effective against hematological malignancies, it has limited efficacy in the treatment of solid tumours [86,87]. Se-containing SAHA derivatives have been developed to overcome the shortfalls of SAHA. The most well-investigated SAHA Se-containing analogue includes the Se-dimer SelSA-1, also known as Bis(5-phenylcarbamoylpentyl) diselenide [B(PCP)−2Se], and the selenocyanide SelSA-2, also known as 5-phenylcarbamoylpentyl selenocyanide (PCP-SeCN), and a ferrocenyl modified SelSA analogue known as Fe-SelSA (Figure 5).
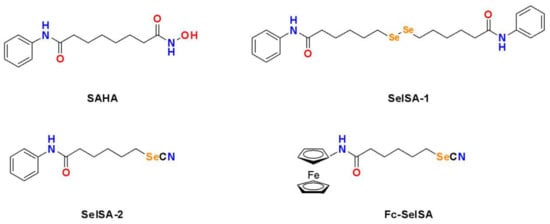
Figure 5. Structures of the HDAC inhibitor SAHA and its selenium-containing derivatives.
SelSa-1 and SelSa-2 were developed in 2010 by Desai and co-workers. Its inhibitory activity was evaluated in Hela nuclear extracts and its effectiveness compared against SAHA. Both SelSA-1 (50 nM) and SelSA-2 (50 nM) were shown to be the superior HDAC inhibitors, disrupting HDAC activity by 81% and 95%, respectively, whereas SAHA (500 nM) only inhibited HDAC activity by 77% [88]. Similar results were observed by Gowda Madhunapantula [89] in Hela nuclear extracts. SelSA-1 or SelSA-2 dose-dependently decreased HDAC activity in the WM35 melanocytic lesion cell line, which resulted in the acetylation of histones H3 and H4. SAHA was 50–60% less effective against obstructing HDAC activity compared to SelSA compounds. Moreover, the topical application of the SAHA Se-derivatives was found to kill melanocytic lesions developed on laboratory-generated skin reconstructs two to four times more effectively than SAHA and decreased tumour development by 87% [89]. SelSA-1 and SelSA-2 were also shown to be more effective against lung cancer cell lines (A549, H2126, H1299, H226, H460, H522, H23, and H441) as they exhibited a lower IC50 than SAHA and more potent inhibition of growth activity was observed using the Se derivatives. However, normal lung epithelial cells showed resistance to the SelSA-1 and SelSA-2, suggesting that these SelSA compounds will be well tolerated as compared to SAHA. While the effect of these SAHA derivatives against HDAC activity was not directly investigated, the anti-proliferative effects are due to the induction of autophagy and inhibition of MAPK and PI3K signalling, which are common occurrences during HDAC inhibition [90].
The mechanism of SAHA’s inhibitory action on class I and II HDACs is through the chelation of zinc (Zn2+) ions present in the active sites of HDACS [91]. Replacing the zinc-binding group (carbonyl and hydroxyl amine group) of SAHA with Se improves its affinity for Zn2+ ions, consequently enhancing its effectiveness as an HDAC inhibitor [92]. In silico, modifications with organoselenium to the zinc-binding group of SAHA resulted in 1726 ligands. Further molecular docking simulations revealed that the five best ligands (CC27, HA27, HB28, IB25, and KA7) had better binding affinity and interactions with Zn2+ ions in inhibited HDACS than SAHA [93]. In silico molecular docking revealed that SelSA-1 shares the same common binding sites on class I HDACs (class I) with SAHA. However, differential binding patterns of Sel-SA-1 with HDAC2 and HDAC8 were observed. For instance, HDAC2 appears to bind similar to SAHA, where the SeH of SelSA-1 binds deeply to HDAC8. For HDAC8, SelSA-1 mimics the binding of trichostatin, which is another potent HDAC inhibitor against different cancers [94].
Docking simulations further established that SelSA-2 selectivity inhibited HDAC6 as SelSA-2 adopted a favourable binding position in the active site of HDAC6 with the selenocyanide group engaging in key hydrogen bonds critical for chelation of Zn2+ ions in the catalytic domain [95]. Hydroxamic acid is able to chelate the Zn2+ ion, which can inhibit HDAC activity [96]. This was confirmed in the breast cancer cell lines MCF-7 and MDA-MB-231 as SelSA-2 selectively inhibited HDAC6, resulting in tubulin acetylation. Moreover, SelSA-2 specifically targeted breast tumours in vivo and improved treatment efficacy with fewer side effects compared to SAHA [95]. Modifications to the cap-linker of SelSA-2 with ferrocenyl (FC-SelSa-2) have also demonstrated effectiveness against MDA-MB-231 cells. Molecular docking analysis showed Fc-SelSA formed new hydrogen-bonding interactions with residues D98 and G151, whereas SAHA and SelSA were unable to do so. Moreover, Fc-SelSA was selectively more potent against MDA-MB-231 cells in comparison to MCF-7 cells, with no toxicity against normal cells. In addition, Fc-SelSA showed a relatively low acute toxicity in vivo and significantly inhibited the growth of triple-negative breast cancer in a xenograft mouse model [97]. Given its high HDAC binding affinity and potent therapeutic effect, selenoderivatives of SAHA serve as a highly promising candidate for targeted cancer therapy with clinical translation potential.
4. Ebselen
Ebselen (C13H9NOSe), first synthesised in 1924, was considered pharmacologically irrelevant until its capability as a potent anti-oxidant was established in 1984 [98,99,100]. Ebselen mimics glutathione peroxidase to detoxify ROS. ROS oxidises the resting state selenol (Ebselon–SeH) to selenenic acid (Ebselon–SeOH), which is subsequently reduced to active selenol by glutathione via a selenenyl sulphide intermediate (ebselen–SeSG) [101]. The anti-oxidant actions of ebselen are also demonstrated through its ability to react with the thioredoxin system, responsible for removing ROS and reactive nitrogen species (RNS) [102]. Ebselen’s antioxidative properties have been widely studied, suggesting that it might possess anti-proliferative and anti-cancer properties through ROS production [103]. These anti-cancer characteristics may also be regulated by the inhibition of quiescin sulfhydryl oxidase 1 (QSO1), an enzyme that enhances growth and tumour cell invasion and alters the composition of the intracellular matrix [104].
To identify potential and novel HDAC inhibitors, two separate studies screened drug and compound libraries approved by the Library of Pharmacologically Active Compounds (LOPAC), US FDA, and National Institutes of Health Clinical Collection compound library [105,106]. In the first study, 1280 compounds were evaluated for potential inhibitory activity against class I and IIa HDACs. Ebselen was identified as one of five compounds with inhibitory action against class I and Iia HDACs and was most effective against HDAC2 [105]. The screening of 1360 compounds from FDA and National Institutes of Health Clinical Collection library against HDACs from subtypes 1 to 11 also found ebselen to exhibit selective HDAC inhibition [106]. Ebselen was shown to selectively inhibit the activity of HDACs 5, 6, 8, and 9 by more than 50%. The HDAC inhibitory action of eleven ebselen analogues was also investigated; This entry focused on the Se-containing ebselen analogue, ebselen oxide (Figure 6). Ebselen oxide was also shown to dose-dependently inhibit HDAC 1, 3, 4, 5, 6, 7, 8, and 9 and increased the potency of HDAC8 inhibition in comparison to ebselen. Unlike other synthetic organoselenium, ebselen and ebselen oxide were shown to effectively inhibit nicotinamide adenine dinucleotide (NAD+)-dependent class III HDACs. Ebselen and ebselen oxide dose-dependently inhibited SIRT1, SIRT2, SIRT3, and SIRT5 activities in biochemical assays. The IC50 values of these three compounds on SIRTs were in the range of 0.3 to 6 μM.
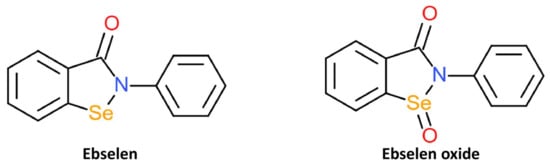
Figure 6. Structures of HDAC inhibitors ebselen and its oxidised derivative, ebselen oxide.
Like MSA, ebselen lacks the characteristic features of HDAC inhibitors to chelate Zn2+ ions present in the active site of HDACs. Its inhibitory action may also be covalent modification to cysteine residues of HDACs, similar to its irreversible inhibitory action against inositol–monophosphatase (IMPase). The therapeutic potential of ebselen is also explored in infectious diseases, such as SARS-CoV-2. A recent study has shown that ebselen and its derivatives inhibit the main protease of SARS-CoV-2 via ebselen interaction with cysteine [107].
This entry is adapted from the peer-reviewed paper 10.3390/ijms222312952
This entry is offline, you can click here to edit this entry!