Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Gastroenterology & Hepatology
Thyroid hormones control lipid metabolism by exhibiting specific effects on the liver and adipose tissue in a coordinated manner. Different diseases of the thyroid gland can result in hypothyroidism. Hypothyroidism is frequently associated with dyslipidemia. Hypothyroidism-associated dyslipidemia subsequently results in intrahepatic accumulation of fat, leading to nonalcoholic fatty liver disease (NAFLD), which leads to the development of hepatic insulin resistance.
- NAFLD
- insulin resistance
- hypothyroidism
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most important chronic liver disease in the western world, affecting almost 30% of the general population. Moreover, the prevalence of NAFLD can be higher in type 2 diabetic patients and obese patients, affecting up to 90% of people with a body mass index higher than 40 kg/m2. NAFLD is also the most rapidly increasing cause of hepatic cirrhosis requiring hepatic transplantation in the future. The pathophysiology of NAFLD is complex and involves multiple hits, but the principal contributing factor to its development is hepatic lipid accumulation, which leads to hepatic insulin resistance [1]. All lipids are not equivalent in terms of their association with the development of insulin resistance. For instance, triglycerides are usually considered inert, whereas diacylglycerols and ceramides can alter insulin signaling [2].
Hypothyroidism can be the result of different diseases of the thyroid gland. Hypothyroidism can be primary, i.e., due to a thyroid gland disorder, or secondary, i.e., due to hypothalamic or pituitary disorders. Primary hypothyroidism is the most frequently encountered in the clinic and can be due to rare congenital disorders (such as thyroid dysgenesis, defective embryonic formation of the gland and genetic diseases) or acquired secondary to different types of thyroiditis (such as Hashimoto’s thyroiditis, silent thyroiditis, subacute thyroiditis and drug-induced thyroiditis) or secondary to surgery or radiotherapy. As thyroid hormones regulate lipid metabolism at various levels in the liver and adipose tissue, hypothyroidism can result in dyslipidemia, which is frequently encountered in hypothyroid patients at the clinic.
2. Thyroid Hormones and Thyroid Hormone Receptors
Thyroid hormones (TH) regulate tissue and cellular metabolism. Tri-iodothyronine (T3) controls gene expression by binding to its receptors. Thyroid hormone receptors (THR) are nuclear receptors, functioning as transcription factors after activation by their ligands [3]. Thyroid hormone receptor isoforms exhibit a tissue-specific expression pattern and function. THR-α (whose gene is located in chromosome 19) mediates TH actions in the heart and brown adipose tissue, whereas, THR-β (whose gene is located in chromosome 3) mediates TH actions on thyroid-stimulating hormone (TSH) secretion and cholesterol metabolism. THR-β has two isoforms. THR-β1 is mainly found in the liver, brain and the kidney, while THR-β2 is found in the hypothalamus and pituitary, exhibiting an important role in the negative feedback of thyroid hormones on the hypothalamic-pituitary axis [4].
Mutations of the THR-β gene are responsible for thyroid hormone resistance syndrome, characterized by tachycardia and increased TSH and free tetra-iodothyronine (FT4) levels.
Thyroid hormone and thyroid receptor agonist treatments have been shown to effectively decrease hepatic steatosis and circulate free fatty acids (FFA) and triglycerides in animal models [5][6]. Research focuses on the beneficial effects of thyroid hormones on metabolism via their receptors while avoiding undesirable effects of systemic hyperthyroidism, such as arrhythmia and bone and muscle loss. Recently, interventional studies have shown the benefits of levothyroxine supplementation in patients with NAFLD and subclinical hypothyroidism in terms of hepatic fat content and liver enzyme levels [7]. Interestingly, these findings were also reproduced in euthyroid individuals with NAFLD [8].
3. Thyroid Hormone Effects on Lipid Metabolism in the Liver and the Adipose Tissue
Thyroid hormones control body weight, lipid and carbohydrate metabolism and thermogenesis. They regulate lipid metabolism by exhibiting specific effects on the liver and adipose tissue, summarized in Figure 1, in a coordinated manner but with occasionally contradictory actions [9].
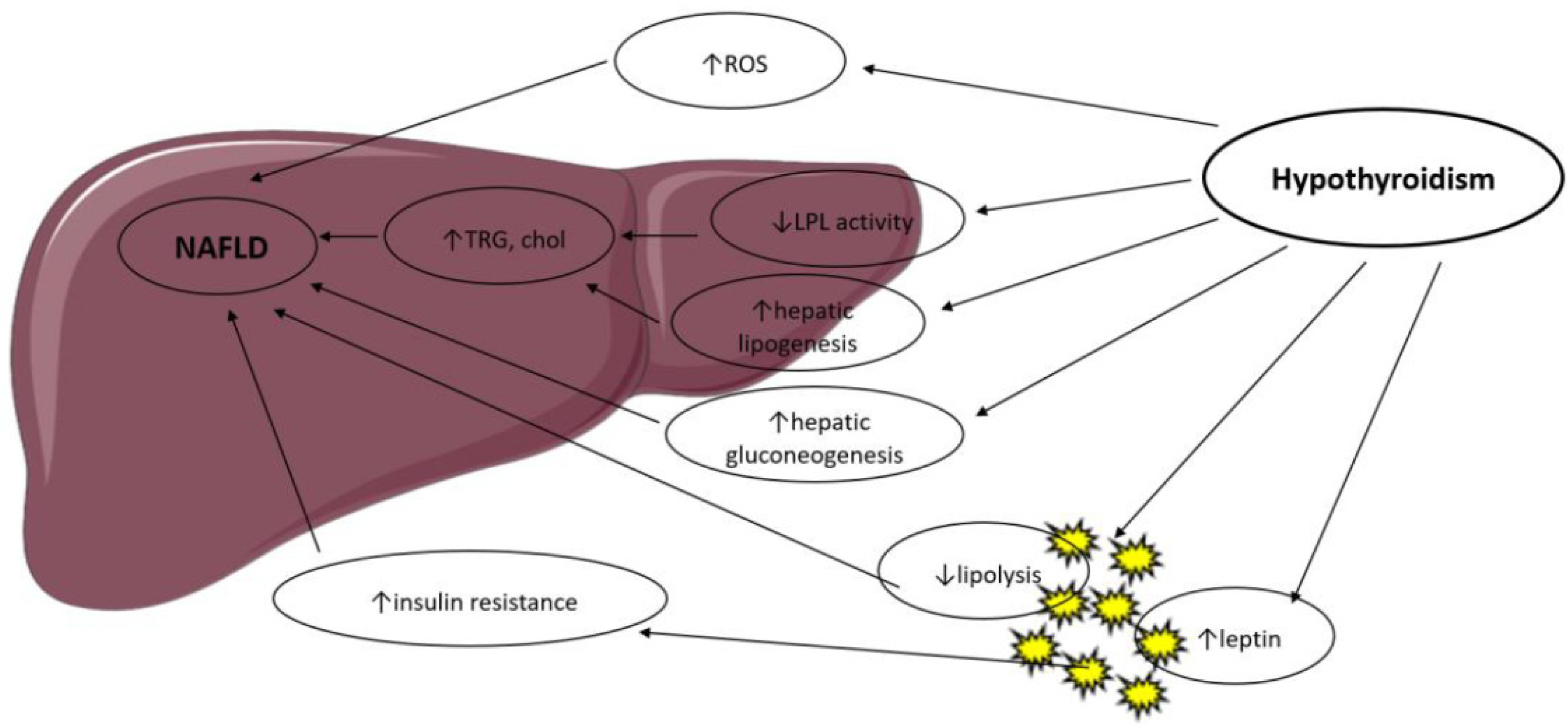
Figure 1. Possible mechanisms in the association between hypothyroidism and NAFLD. LPL: Lipoprotein Lipase; ROS: Reactive Oxygen Species; TRG: Triglyceride; Chol: Cholesterol.
T3 controls the expression of genes involved in hepatic lipogenesis and genes involved in the oxidation of free fatty acids through the thyroid hormone receptor-β, which is the main isoform expressed in the liver [3][10]. Thyroid hormone receptor-α is the main mediator of thyroid hormone actions in the heart and brown adipose tissue. Thus, thyroid hormones regulate lipid metabolism in a tissue-dependent manner, and this was confirmed by studies in knockout mice. THR-α-knockout mice exhibit decreased liver fat content and white adipose tissue mass via a decrease in genes involved in lipogenesis. They have less insulin resistance and hepatic steatosis [11]. THR-β-knockout animals display an increased liver mass and hepatic lipid accumulation through increased lipogenic genes and decreased fatty acid β-oxidation but no significant change in white adipose tissue [11].
Hyperthyroidism has been shown to increase adipose tissue lipolysis [12] and hepatic lipogenesis and is associated with lower body weight, notably due to increased catabolism [13]. These actions are mediated by a T3-induced increase in the expression of several lipogenic genes (such as acyl-CoA-synthetase, fatty acid synthase, acetyl-CoA carboxylase and glucose-6-P dehydrogenase) and genes involved in fatty acid oxidation (such as lipoprotein lipase, fatty acid-binding protein and fatty acid transporter) [3].
Hypothyroidism reduces liver uptake of FFA derived from triglycerides [14] and is associated with a decrease in lipolysis in the adipose tissue and decreased cholesterol clearance [15]. As a result, β-oxidation of free fatty acids and triglyceride clearance is reduced, with a consequent hepatic accumulation of triglycerides and increased low-density lipoprotein (LDL) uptake. Hypothyroidism reduces hepatic lipase activity, which mediates fatty acid oxidation and long-chain fatty acids’ oxidation for energy production. Lipid storage in the liver is further increased by obesity and low resting energy expenditure, both enhanced by hypothyroidism [16][17]. Thyroid hormone treatment in human and murine models reverses hepatic lipase reduced activity.
In the mitochondria, thyroid hormones stimulate carnitine palmitoyltransferase-1a (Cpt1a), the rate-limiting enzyme in fatty-acid oxidation.
Obesity, in both human and animal studies, is found to lead to lipid accumulation in the liver, resulting in fibrosis and cirrhosis. Increased hepatic lipid deposition induces downregulation of several metabolism-related genes, which are dependent of T3 actions [3].
Thyroid hormones are activators of lipogenesis through direct and indirect mechanisms. T3 stimulates enzymes that catalyze several important steps of hepatic fatty acid synthesis, such as acetyl-CoA carboxylase (which catalyzes the carboxylation of acetyl-CoA to malonyl-CoA, the first step of hepatic fatty acid synthesis) and fatty acid synthetase [18]. T3 also induces several transcription factors that participate in de novo lipogenesis, such as carbohydrate responsive element-binding protein (ChREBP), a strong lipogenic regulator [19]. Thyroid-stimulating hormone is also believed to stimulate hepatic lipogenesis through binding with the TSH-receptor expressed at the surface of the hepatocytes, which further leads to stimulation of the peroxisome proliferator-activated receptor-α (PPARα) pathway and activation of sterol regulatory element-binding transcription factor 1 (SREBP-1c) [20][21]. TSH directly increases hepatic gluconeogenesis and decreases phosphorylation of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the main target of statins, thereby inducing hypercholesterolemia [22].
Oxidative stress derived from β-oxidation is thought to contribute to the progression of nonalcoholic steatohepatitis (NASH) to hepatocyte inflammation and liver fibrosis. Hyperthyroidism has been shown to increase oxidative stress, leading to liver cell injury [23], while hypothyroidism lowers oxidative stress levels through a decrease in energy expenditure [24]. Thus, thyroid hormones may contribute to the progression of nonalcoholic fatty liver disease to nonalcoholic steatohepatitis, but the exact pathophysiological mechanisms remain to be clarified.
4. Thyroid Hormones and Dyslipidemia
Hypothyroidism is associated with hyperlipidemia through modifications in lipid synthesis, absorption, circulation and metabolism. Thyroid hormones increase cholesterol synthesis by increasing the expression of HMG-CoA reductase in the liver [25]. Thus, hypothyroidism leads to decreased hepatic cholesterol synthesis. However, two additional concomitant mechanisms outweighed this effect. First, there is an increase in gastro-intestinal cholesterol absorption mediated by the Niemann-Pick C1-like 1 protein, the target of the lipid-lowering molecule ezetimibe, in the gut [26]. Second, there is a decrease in cell-surface LDL-cholesterol receptors, possibly via T3-mediated effects on the sterol regulatory element-binding protein-2 (SREBP-2), leading to reduced plasma LDL-cholesterol clearance and increased apo-B lipoproteins [27].
Hypothyroidism also decreases cholesterol excretion and plasma triglyceride clearance, the latter through a decrease in lipoprotein lipase levels [28]. Plasma cholesteryl ester transfer proteins (CETPs), shifting cholesterol from high-density lipoproteins (HDL-C) to LDL-C and very low-density lipoproteins (VLDL) are reduced in hypothyroid states [29].
The combined result of the above changes is an increase in total cholesterol and LDL levels, a slight increase in HDL and triglycerides levels and triglyceride accumulation in the liver, a risk factor for the development of nonalcoholic fatty liver disease [30]. Increased triglyceride accumulation in the liver also contributes to the development of hepatic insulin resistance, another condition linking hypothyroidism with NAFLD, which will be discussed later.
Observational studies confirm that among patients with overt hypothyroidism, 30% have increased total cholesterol and LDL levels, and 90% have dyslipidemia. Furthermore, levothyroxine treatment reverses lipid alterations, with the exception of patients with underlying hyperlipidemia [31].
The effect of subclinical hypothyroidism on lipid levels is less obvious, and the results of clinical studies have been inconsistent. Some observational studies found no difference in lipid levels among subclinical hypothyroid patients and matched controls [32][33], whereas others found significantly higher total cholesterol, triglycerides and LDL-C levels in subclinical hypothyroidism [34][35]. Insulin resistance and smoking are believed to be possible confounding factors since they both induce higher cholesterol increase in the presence of hypothyroidism [31].
5. NAFLD and Dysregulated Lipid Metabolism
The pathophysiology of NAFLD is complex, multifactorial and involves multiple systemic alterations [36]. The classical “two-hit” theory is divided into a first “hit” with intrahepatic accumulation of fatty acids and a second “hit” that includes other factors such as oxidative stress and mitochondrial dysfunction. Nevertheless, this theory has been considered inadequate to fully represent the pathogenesis of NAFLD. Therefore, it has been replaced by the “multiple parallel hits” hypothesis that more accurately represents the process of NAFLD development and progression. Indeed, various factors, such as genetic and environmental factors (notably dietary habits), act in parallel and in a synergic way to cause NAFLD [36][37]. NAFLD is due to hepatic lipid accumulation that will subsequently lead to hepatic insulin resistance, alterations in gut microbiota and other deleterious phenomena such as mitochondrial dysfunction, endoplasmic reticulum stress, oxidative stress and production of reactive oxygen species [38]. These different deleterious elements will subsequently lead to a chronic inflammatory state in the liver, promoting NAFLD and NASH [39].
Hepatic lipid accumulation consists of different lipid intermediates, such as triglycerides, which are usually considered inert, and diacylglycerols and ceramides, which have been shown to cause hepatic insulin resistance in different animal models of nonalcoholic fatty liver disease [40][41][42][43][44][45][46]. Insulin resistance also promotes hepatic de novo lipogenesis and adipose tissue lipolysis, leading to an increased flux of free fatty acids to the liver [47]. This process is also associated with an increase in plasma triglycerides (TG) concentration and a reduction in plasma HDL concentration, contributing to the atherogenic dyslipidemia seen in NAFLD [48]. The plasma HDL level is usually lower in insulin-resistant states, which can be explained by the following mechanism: VLDL TG can be exchanged for HDL cholesterol in the presence of increased plasma VLDL concentrations and the normal activity of cholesteryl ester transfer protein, where a VLDL particle gives a molecule of TG to an HDL particle in return for one of the cholesteryl ester molecules from HDL. This mechanism leads to a cholesterol-rich VLDL remnant particle that is atherogenic and a TG rich, cholesterol-depleted HDL particle [49]. The TG-rich HDL particle will then undergo further change, notably hydrolysis of its TG, which will lead to the dissociation of the apoA-1 protein. Subsequently, the free apoA-1 will be cleared more rapidly in the plasma than the apoA-1 bound to HDL particles, and this process results in reduced circulating apoA-1, HDL cholesterol and the absolute number of HDL particles [50], as summarized in Figure 2. Altogether, these processes lead to the dysregulated lipid metabolism seen in NAFLD.
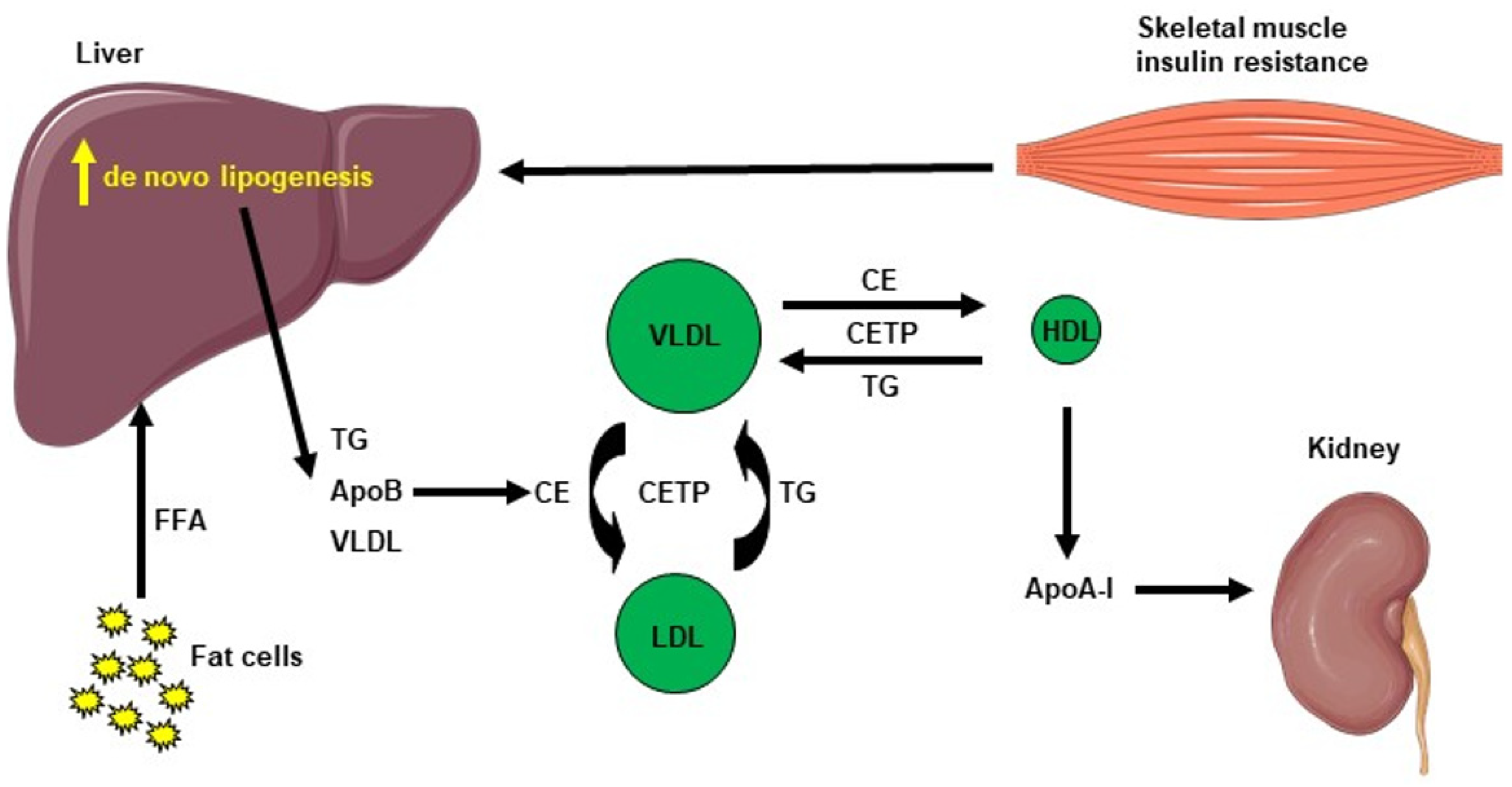
Figure 2. Cholesterol metabolism induced by hepatic de novo lipogenesis. Skeletal muscle insulin resistance increases hepatic de novo lipogenesis, leading to increased hepatic triglycerides (TG). TG can be exchanged for high-density lipoprotein (HDL) cholesterol in the presence of increased plasma very low–density lipoprotein (VLDL) concentrations and normal activity of cholesteryl ester transfer protein (CETP). A VLDL particle then donates a molecule of TG to an HDL particle in return for one of the cholesteryl ester (CE) molecules from HDL. The TG-rich HDL particle can be hydrolyzed of its TG, leading to dissociation of the Apolipoprotein A-1 (Apo A-1) protein. The resulting free Apo A-1 is cleared more rapidly in plasma than the apo A-1 bound to HDL particles, leading to reduced circulating apo A-1, HDL cholesterol and the number of HDL particles.
This entry is adapted from the peer-reviewed paper 10.3390/ijms222312797
References
- Tanase, D.M.; Gosav, E.M.; Neculae, E.; Costea, C.F.; Ciocoiu, M.; Hurjui, L.L.; Tarniceriu, C.C.; Floria, M. Hypothyroidism-Induced Nonalcoholic Fatty Liver Disease (HIN): Mechanisms and Emerging Therapeutic Options. Int. J. Mol. Sci. 2020, 21, 5927.
- Jornayvaz, F.R.; Shulman, G.I. Diacylglycerol activation of protein kinase Cepsilon and hepatic insulin resistance. Cell Metab. 2012, 15, 574–584.
- Huang, Y.Y.; Gusdon, A.M.; Qu, S. Cross-talk between the thyroid and liver: A new target for nonalcoholic fatty liver disease treatment. World J. Gastroenterol. 2013, 19, 8238–8246.
- Motomura, K.; Brent, G.A. Mechanisms of thyroid hormone action. Implications for the clinical manifestation of thyrotoxicosis. Endocrinol. Metab. Clin. N. Am. 1998, 27, 1–23.
- Perra, A.; Simbula, G.; Simbula, M.; Pibiri, M.; Kowalik, M.A.; Sulas, P.; Cocco, M.T.; Ledda-Columbano, G.M.; Columbano, A. Thyroid hormone (T3) and TRbeta agonist GC-1 inhibit/reverse nonalcoholic fatty liver in rats. FASEB J. 2008, 22, 2981–2989.
- Cable, E.E.; Finn, P.D.; Stebbins, J.W.; Hou, J.; Ito, B.R.; van Poelje, P.D.; Linemeyer, D.L.; Erion, M.D. Reduction of hepatic steatosis in rats and mice after treatment with a liver-targeted thyroid hormone receptor agonist. Hepatology 2009, 49, 407–417.
- Liu, L.; Yu, Y.; Zhao, M.; Zheng, D.; Zhang, X.; Guan, Q.; Xu, C.; Gao, L.; Zhao, J.; Zhang, H. Benefits of Levothyroxine Replacement Therapy on Nonalcoholic Fatty Liver Disease in Subclinical Hypothyroidism Patients. Int. J. Endocrinol. 2017, 2017, 5753039.
- Bruinstroop, E.; Dalan, R.; Cao, Y.; Bee, Y.M.; Chandran, K.; Cho, L.W.; Soh, S.B.; Teo, E.K.; Toh, S.A.; Leow, M.K.S.; et al. Low-Dose Levothyroxine Reduces Intrahepatic Lipid Content in Patients With Type 2 Diabetes Mellitus and NAFLD. J. Clin. Endocrinol. Metab. 2018, 103, 2698–2706.
- Sinha, R.A.; You, S.H.; Zhou, J.; Siddique, M.M.; Bay, B.H.; Zhu, X.; Privalsky, M.L.; Cheng, S.Y.; Stevens, R.D.; Summers, S.A.; et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J. Clin. Investig. 2012, 122, 2428–2438.
- Flores-Morales, A.; Gullberg, H.; Fernandez, L.; Stahlberg, N.; Lee, N.H.; Vennstrom, B.; Norstedt, G. Patterns of liver gene expression governed by TRbeta. Mol. Endocrinol. 2002, 16, 1257–1268.
- Jornayvaz, F.R.; Lee, H.Y.; Jurczak, M.J.; Alves, T.C.; Guebre-Egziabher, F.; Guigni, B.A.; Zhang, D.; Samuel, V.T.; Silva, J.E.; Shulman, G.I. Thyroid hormone receptor-alpha gene knockout mice are protected from diet-induced hepatic insulin resistance. Endocrinology 2012, 153, 583–591.
- Riis, A.L.; Gravholt, C.H.; Djurhuus, C.B.; Norrelund, H.; Jorgensen, J.O.; Weeke, J.; Moller, N. Elevated regional lipolysis in hyperthyroidism. J. Clin. Endocrinol. Metab. 2002, 87, 4747–4753.
- Cachefo, A.; Boucher, P.; Vidon, C.; Dusserre, E.; Diraison, F.; Beylot, M. Hepatic lipogenesis and cholesterol synthesis in hyperthyroid patients. J. Clin. Endocrinol. Metab. 2001, 86, 5353–5357.
- Klieverik, L.P.; Coomans, C.P.; Endert, E.; Sauerwein, H.P.; Havekes, L.M.; Voshol, P.J.; Rensen, P.C.; Romijn, J.A.; Kalsbeek, A.; Fliers, E. Thyroid hormone effects on whole-body energy homeostasis and tissue-specific fatty acid uptake in vivo. Endocrinology 2009, 150, 5639–5648.
- Nedvidkova, J.; Haluzik, M.; Bartak, V.; Dostalova, I.; Vlcek, P.; Racek, P.; Taus, M.; Behanova, M.; Svacina, S.; Alesci, S.; et al. Changes of noradrenergic activity and lipolysis in the subcutaneous abdominal adipose tissue of hypo- and hyperthyroid patients: An in vivo microdialysis study. Ann. N. Y. Acad. Sci. 2004, 1018, 541–549.
- D’Ambrosio, R.; Campi, I.; Maggioni, M.; Perbellini, R.; Giammona, E.; Stucchi, R.; Borghi, M.; Degasperi, E.; De Silvestri, A.; Persani, L.; et al. The relationship between liver histology and thyroid function tests in patients with non-alcoholic fatty liver disease (NAFLD). PLoS ONE 2021, 16, e0249614.
- Hazlehurst, J.M.; Tomlinson, J.W. Non-alcoholic fatty liver disease in common endocrine disorders. Eur. J. Endocrinol. 2013, 169, R27–R37.
- Yin, L.; Zhang, Y.; Hillgartner, F.B. Sterol regulatory element-binding protein-1 interacts with the nuclear thyroid hormone receptor to enhance acetyl-CoA carboxylase-alpha transcription in hepatocytes. J. Biol. Chem. 2002, 277, 19554–19565.
- Dentin, R.; Girard, J.; Postic, C. Carbohydrate responsive element binding protein (ChREBP) and sterol regulatory element binding protein-1c (SREBP-1c): Two key regulators of glucose metabolism and lipid synthesis in liver. Biochimie 2005, 87, 81–86.
- Gariani, K.; Jornayvaz, F.R. Pathophysiology of NASH in endocrine diseases. Endocr. Connect. 2021, 10, R52–R65.
- Ritter, M.J.; Amano, I.; Hollenberg, A.N. Thyroid Hormone Signaling and the Liver. Hepatology 2020, 72, 742–752.
- Li, Y.; Wang, L.; Zhou, L.; Song, Y.; Ma, S.; Yu, C.; Zhao, J.; Xu, C.; Gao, L. Thyroid stimulating hormone increases hepatic gluconeogenesis via CRTC2. Mol. Cell. Endocrinol. 2017, 446, 70–80.
- Messarah, M.; Boumendjel, A.; Chouabia, A.; Klibet, F.; Abdennour, C.; Boulakoud, M.S.; Feki, A.E. Influence of thyroid dysfunction on liver lipid peroxidation and antioxidant status in experimental rats. Exp. Toxicol. Pathol. 2010, 62, 301–310.
- Loria, P.; Carulli, L.; Bertolotti, M.; Lonardo, A. Endocrine and liver interaction: The role of endocrine pathways in NASH. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 236–247.
- Choi, J.W.; Choi, H.S. The regulatory effects of thyroid hormone on the activity of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Endocr. Res. 2000, 26, 1–21.
- Galman, C.; Bonde, Y.; Matasconi, M.; Angelin, B.; Rudling, M. Dramatically increased intestinal absorption of cholesterol following hypophysectomy is normalized by thyroid hormone. Gastroenterology 2008, 134, 1127–1136.
- Shin, D.J.; Osborne, T.F. Thyroid hormone regulation and cholesterol metabolism are connected through Sterol Regulatory Element-Binding Protein-2 (SREBP-2). J. Biol. Chem. 2003, 278, 34114–34118.
- Lam, K.S.; Chan, M.K.; Yeung, R.T. High-density lipoprotein cholesterol, hepatic lipase and lipoprotein lipase activities in thyroid dysfunction--effects of treatment. Q. J. Med. 1986, 59, 513–521.
- Tan, K.C.; Shiu, S.W.; Kung, A.W. Plasma cholesteryl ester transfer protein activity in hyper- and hypothyroidism. J. Clin. Endocrinol. Metab. 1998, 83, 140–143.
- Duntas, L.H. Thyroid disease and lipids. Thyroid 2002, 12, 287–293.
- Pearce, E.N. Update in lipid alterations in subclinical hypothyroidism. J. Clin. Endocrinol. Metab. 2012, 97, 326–333.
- Vierhapper, H.; Nardi, A.; Grosser, P.; Raber, W.; Gessl, A. Low-density lipoprotein cholesterol in subclinical hypothyroidism. Thyroid 2000, 10, 981–984.
- Hueston, W.J.; Pearson, W.S. Subclinical hypothyroidism and the risk of hypercholesterolemia. Ann. Fam. Med. 2004, 2, 351–355.
- Kanaya, A.M.; Harris, F.; Volpato, S.; Perez-Stable, E.J.; Harris, T.; Bauer, D.C. Association between thyroid dysfunction and total cholesterol level in an older biracial population: The health, aging and body composition study. Arch. Intern. Med. 2002, 162, 773–779.
- Canaris, G.J.; Manowitz, N.R.; Mayor, G.; Ridgway, E.C. The Colorado thyroid disease prevalence study. Arch. Intern. Med. 2000, 160, 526–534.
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol. Commun. 2020, 4, 478–492.
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048.
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647.
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377.
- Asrih, M.; Altirriba, J.; Rohner-Jeanrenaud, F.; Jornayvaz, F.R. Ketogenic Diet Impairs FGF21 Signaling and Promotes Differential Inflammatory Responses in the Liver and White Adipose Tissue. PLoS ONE 2015, 10, e0126364.
- Camporez, J.P.; Asrih, M.; Zhang, D.; Kahn, M.; Samuel, V.T.; Jurczak, M.J.; Jornayvaz, F.R. Hepatic insulin resistance and increased hepatic glucose production in mice lacking Fgf21. J. Endocrinol. 2015, 226, 207–217.
- Camporez, J.P.G.; Kanda, S.; Petersen, M.C.; Jornayvaz, F.R.; Samuel, V.T.; Bhanot, S.; Petersen, K.F.; Jurczak, M.J.; Shulman, G.I. ApoA5 knockdown improves whole-body insulin sensitivity in high-fat-fed mice by reducing ectopic lipid content. J. Lipid Res. 2015, 56, 526–536.
- Somm, E.; Montandon, S.A.; Loizides-Mangold, U.; Gaia, N.; Lazarevic, V.; De Vito, C.; Perroud, E.; Bochaton-Piallat, M.L.; Dibner, C.; Schrenzel, J.; et al. The GLP-1R agonist liraglutide limits hepatic lipotoxicity and inflammatory response in mice fed a methionine-choline deficient diet. Transl. Res. 2021, 227, 75–88.
- Montandon, S.A.; Somm, E.; Loizides-Mangold, U.; de Vito, C.; Dibner, C.; Jornayvaz, F.R. Multi-technique comparison of atherogenic and MCD NASH models highlights changes in sphingolipid metabolism. Sci. Rep. 2019, 9, 16810.
- Somm, E.; Jornayvaz, F.R. Fibroblast Growth Factor 15/19: From Basic Functions to Therapeutic Perspectives. Endocr. Rev. 2018, 39, 960–989.
- Somm, E.; Henry, H.; Bruce, S.J.; Aeby, S.; Rosikiewicz, M.; Sykiotis, G.P.; Asrih, M.; Jornayvaz, F.R.; Denechaud, P.D.; Albrecht, U.; et al. beta-Klotho deficiency protects against obesity through a crosstalk between liver, microbiota, and brown adipose tissue. JCI Insight 2017, 2, e91809.
- Bugianesi, E.; Moscatiello, S.; Ciaravella, M.F.; Marchesini, G. Insulin resistance in nonalcoholic fatty liver disease. Curr. Pharm. Des. 2010, 16, 1941–1951.
- Jornayvaz, F.R.; Samuel, V.T.; Shulman, G.I. The role of muscle insulin resistance in the pathogenesis of atherogenic dyslipidemia and nonalcoholic fatty liver disease associated with the metabolic syndrome. Annu. Rev. Nutr. 2010, 30, 273–290.
- Krauss, R.M.; Siri, P.W. Metabolic abnormalities: Triglyceride and low-density lipoprotein. Endocrinol. Metab. Clin. N. Am. 2004, 33, 405–415.
- Ginsberg, H.N. Insulin resistance and cardiovascular disease. J. Clin. Investig. 2000, 106, 453–458.
This entry is offline, you can click here to edit this entry!