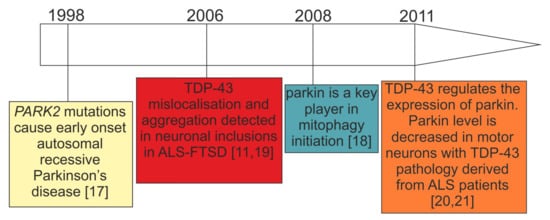
4.1. Consistent Effects of TDP-43 Depletion on Parkin Levels
At the mechanistic level of RNA processing, TDP-43 has turned out to be crucial for the maintenance of brain enriched mRNAs with long introns (>100 kb) such as
PARK2 [
20,
21]. Indeed,
PARK2 pre-mRNA possesses multiple TDP-43 binding sites, suggesting that the control of its stability depends on TDP-43 RNA binding function, at least partially [
20] (
Figure 2C). In keeping with this view, and irrespectively of animal or cellular models under investigation, TDP-43 depletion consistently resulted in parkin mRNA/protein downregulation [
20,
21,
91,
94] (
Table 1).
To date, a decrease in parkin mRNA has been observed upon TDP-43 depletion in mouse adult brain, stem cell-derived human neurons, HEK293T cells, human primary skin fibroblasts, and motor neurons obtained from patients with sporadic ALS [
20,
21,
91,
94] (
Table 1). However, in motor neurons derived from patients with sporadic ALS, parkin decrease correlated with the presence of TDP-43 aggregates while ca. 95% of motor neurons without TDP-43 pathology showed normal parkin levels [
21].
Recently, Sun et al., have elegantly shown that TDP-43 can affect parkin expression post-transcriptionally and in an intron/UTR-independent manner [
92]. In a recent study, the authors have observed that overexpression of human TDP-43 in HEK293T cells downregulated plasmid-expressed, intron-free parkin, both at the mRNA and protein level (
Table 1). This required both the RNA-binding and protein–protein interaction domains of TDP-43 that included the RNA recognition motif 1 (RRM1) and the glycine-rich domain (GRD) domain in the C-terminus [
92].
In conclusion, the consistency of the results of TDP-43 depletion (Table 1) in different animal and cellular models and in patients with ALS/FTLD suggests that TDP-43 loss-of-function is crucial to maintaining parkin expression levels. Whether this is the only mechanism by which parkin decreases in ALS/FTLD remains elusive.
4.2. Discrepant Effects of TDP-43 Overexpression on Parkin Levels
Supporting the notion of parkin being a direct target for TDP-43, a significant increase in
PARK2 mRNA was observed in transgenic mice (hTDP43-Tg) brains compared to controls and a few cellular TDP-43 overexpression models [
91,
93,
94]. Transgenic hemizygous mice harboring human TDP-43 A315T had increased levels of both soluble and insoluble parkin (by ca. 50 and 60%, respectively) [
93] (
Table 1). In contrast, other groups reported parkin downregulation upon wild-type and mutant TDP-43 overexpression, depending on the cellular model used [
92,
94,
96] (
Table 1).
Apart from parkin, TDP-43 ectopic expression influenced also other mitophagy key players. Sun et al. reported accumulation of cleaved PINK1(~52 kDa) insoluble aggregates in the cytoplasm via the mechanism of TDP-43 overexpression–related impairment of the proteasomal activity [
92]. To confirm that these findings hold true also in vivo, the authors confirmed aggregation of cleaved endogenous PINK1 in the motor cortex of mice expressing Q331K mutant of TDP-43 [
92]. Finally, they demonstrated that TDP-43-driven PINK1 accumulation affected negatively mitochondrial respiratory functions and lifespan in the
Drosophila model [
92].
At present, there is no satisfactory explanation for the observed differences between TDP-43 overexpression experiments. In contrast to TDP-43 knockdown, it should nonetheless be considered that TDP-43 overexpression levels may vary substantially between different labs/experimental systems. At the cellular level, this may result in the overexpressed TDP-43 binding/interacting with different partners, depending on the absolute level of overexpression reached during the study. If this could be the case, it would not be surprising that different overexpression levels could even have opposite effects on parkin expression. Of course, such a possibility would have to be experimentally tested and this is an area that certainly deserves further investigation.
4.4. Unanswered Question 1: Is Parkin Downregulation in TDP-43 Proteinopathies Functionally Relevant (Molecular Biology Perspective)?
While biallelic
PARK2 mutations lead to complete parkin depletion in patients with PD [
98,
99] (
Figure 2A), the parkin decrease in carriers of single
PARK2 mutations remains yet to be determined (
Figure 2B). In fact, it is important to note that in cellular PD models almost complete parkin deficiency (ca. 80%), obtained by silencing with small interfering RNAs (siRNA), is sufficient to trigger mitochondrial dysfunction in wild-type fibroblasts [
100]. However, in contrast to patients with EOPD due to homozygous
PARK2 mutations, healthy carriers of heterozygous parkin mutation do not present abnormal mitochondrial function, including deregulated ultrastructure, morphology, and metabolism [
99,
101]. Likewise, 50% parkin silencing, which models haploinsufficiency (that would correspond to single
PARK2 mutation carriers), did not change any mitochondrial parameters, thus indicating the importance of the dose [
100].
How do the above-mentioned observations correspond to TDP-43 proteinopathies? First of all, many transcriptomic/proteomic analyses in patients with ALS–FTSD or TDP-43 proteinopathy animal models do not report parkin expression alterations [
102,
103,
104,
105,
106]. These might be due to the fact that all “omic” results (RNAseq, microarrays, and proteomics) carry a bias, since they are not able to distinguish between healthy cells and those with TDP-43 pathology. Moreover, the severity of TDP-43 pathology may be of varying grades [
107,
108].
In models where parkin expression changes are observed (
Section 5.1 and
Table 1),
PARK2 mRNA decrease upon TDP-43 depletion ranges from c.a. 25% in human neurons, 60% in human fibroblasts to 60–80% in rodent models. However, TDP-43 depletion does not mirror the complex pathology present in the brains of patients with ALS–FTLD. Thus, the local threshold effect might be extremely important, in terms of neurons with TDP-43 pathology and magnitude of parkin decrease. Furthermore, additional genetic or epigenetic risk factors may decide to what extent mitochondrial dysfunction would manifest itself.
Since carriers of single
PARK2 mutations can present subclinical brain dysfunction (see
Section 3.2 for detailed description) it could therefore be speculated that subtle parkin decrease in TDP-43 proteinopathies can lead to similar subclinical phenotypes as in single
PARK2 mutation carriers. Nonetheless, this is an assumption that will need careful validation in the future. Indeed, there are limited existing data regarding parkinsonian phenotypes observed in TDP-43 proteinopathies, and this subject is discussed in
Section 5.5 below.
4.5. Unanswered Question 2: What Is the Evidence of Parkinsonism in TDP-43 Proteinopathies (Clinical Perspective)?
ALS–FTSD comprises cases with dementia (ALS–FTD), cases with behavioral and/or cognitive impairment without dementia (ALSbci, ALSbi, and ALSci), ALS–parkinsonism/dementia complex (ALS–PDC, named also Western Pacific variant of ALS, lytico bodig), and other mixed variants [
9].
ALS–FTSD is predominantly associated with TDP-43 proteinopathy [
109], ALS being the most common one, and includes many movement disorders presentations, but MND and parkinsonian symptoms predominate.
First of all, it is important to keep in mind that cases of ALS–FTSD that have not been genetically defined may present other types of pathology (such as TAU, FUS, etc.). For this reason, in this review we focus on parkinsonism in genetic forms of ALS–FTSD with confirmed TDP-43 proteinopathy, caused by mutations in
C9orf72,
PGRN, and
TARDBP [
110,
111,
112,
113].
Regarding FTLD it is interesting to note that the first links between this disease and parkinsonism extend before the recognition of TDP-43 proteinopathies. Indeed, until a few decades ago parkinsonian symptoms were regarded as a rare manifestation of FTLD associated with
MAPT mutation, so the term frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) was coined [
114]. More recently, the association between progranulin mutations and parkinsonism [
115,
116,
117] enabled us to distinguish between FTDP-17 (MAPT) and FTDP-17 (PGRN) [
117]. Currently, the FTDP-17 term is no longer used clinically, considering that parkinsonism in FTLD has been shown to extend far beyond mutations in chromosome 17 [
118].
Usually, FTLD is associated with either parkinsonian symptoms (e.g., in PGRN mutations) or with MND (e.g., in C9orf72).
In genetic FTLD parkinsonism is the most frequent movement disorder manifestation. It occurs in about 80% of cases. However, only ~ 1 in 3 cases with
C9orf72 mutation and 1 in 10 cases with
PGRN mutations present with parkinsonism at onset [
119]. According to other sources, depending on the population, parkinsonian symptoms occur in >20% of patients with ALS linked to
C9orf72 mutation, and as much as 50% to 75% of cases with bvFTD [
120,
121].
PGRN mutations are associated predominantly with parkinsonism and only occasionally with MND [
122,
123]. In some
PGRN kindreds, parkinsonism has been reported in up to 80% of cases [
124]. Importantly, patients with
PGRN mutations demonstrate presynaptic dopaminergic deficit, as evidenced by Dopamine Transporter Scan (DAT-Scan) [
125]. Unfortunately, DAT-Scan is not routinely performed, and parkinsonism in FTLD is usually described only based on clinical presentation.
It seems that
TARDBP mutations may have a broader symptomatic spectrum than other gene mutations in FTLD, as they may be associated with all three conditions: ALS, FTLD, and parkinsonism [
126].
At the clinical level, parkinsonism in FTLD syndromes is typically characterized by akinetic–rigid phenotype (symmetrical muscle rigidity, bradykinesia, hypokinesia, parkinsonian gait, and rarely resting tremor) [
127] and may share clinical features with either progressive supranuclear palsy (PSP) or corticobasal syndrome (CBS) [
128,
129]. Sometimes multiple system atrophy-like presentations occur with dysautonomia, ataxia, and pyramidal symptoms [
127].
Akinetic–rigid parkinsonism, as far as TDP-43 proteinopathies are concerned, occurs commonly in
PGRN mutations and is rather uncommon in
C9orf72 mutations and
VCP (Valosin Containing Protein) mutations and rare in
TARDP mutations cases. PSP-like features may be rarely observed in both
C9orf72 and
PGRN mutation carriers, CBS-like features are uncommon in
PGRN and rare in
C9orf72 [
127]. Of note, parkinsonian symptoms may be the first symptom in FTLD or develop after the occurrence of language or behavioral symptoms [
121,
130].
Among inherited FTLD cases, MND is observed mainly in patients carrying C9orf72 or TARDBP gene mutations, but also those with DCTN1 (Dynactin Subunit 1) and VCP gene mutations.
In some very rare clinical entities, such as Perry disease, caused by
DCTN1 mutation, TDP-43 pathology is predominant [
131]. In others, it co-occurs with tau pathology (PSP, CBD) or alpha-synuclein pathology (PD, DLB) [
13]. In Perry disease, parkinsonism, central hypoventilation, and weight loss [
131] are accompanied by behavioral manifestations. The syndrome shares symptoms of PD, ALS and may fall into the FTLD spectrum [
132].
Another example of the complex relationship between ALS and parkinsonism is the rare variant of ALS: ALS and parkinsonism/dementia complex (ALS/PDC) in which TDP-43 pathology may be accompanied by alpha-synuclein pathology. In ALS/PDC three types of pathology were described: the tauopathy-dominant type, the TDP-43 proteinopathy-dominant type, and the synucleinopathy-dominant type [
133].
The frequency of parkinsonism in ALS–FTSD with TDP-43 proteinopathy cannot be easily established. Parkinsonism may be an under-diagnosed phenomenon in ALS–FTSD with TDP-43 proteinopathy due to several reasons. First, genetic testing and/or neuropathological examination is not routinely performed worldwide, remaining unproven in many cases. Secondly,
C9orf72 is a relatively recently described mutation as are Strong et al.’s criteria [
9]. Third, at the diagnostic stage patients usually attend either a Dementia Clinic or Movement Disorders Clinics. Inevitably, some clinics focus mainly on cognitive/behavioral symptoms or motor symptoms and a mixed presentation of ALS–FTSD may be overlooked, especially if parkinsonism is not present at onset and develops later. At more advanced disease stages when the patients require constant supervision/care they are rarely seen by movement disorder specialists. Therefore, parkinsonism may not be diagnosed even in cases with pronounced akinetic–rigid manifestations. Furthermore, since akinetic–rigid parkinsonism is not a commonly known presentation of parkinsonism, specialists without movement disorder expertise are less likely to diagnose correctly someone that does not present with tremor phenotype.
In conclusion, the theme of parkinsonism in ALS–FTLD guarantees further research since at present there are a lot of gaps that need to be filled, specifically regarding parkinsonism in sporadic forms of ALS–FTSD with defined neuropathology and its clinical subgroups. Finally, it would be interesting to investigate whether parkinsonian symptoms observed in ALS–FTLD could be mediated by TDP-43-associated parkin decrease.