Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Mercury is a heavy metal found in organic and inorganic forms that represents an important toxicant with impact on human health. Mercury can be released in the environment by natural phenoms (i.e., volcanic eruptions), industrial products, waste, or anthropogenic actions (i.e., mining activity).
- mercury
- ethanol
- depression
- anxiety
- insomnia
- pollutant
1. Introduction
Exposure to environmental toxicants has impact on human health worldwide. Mercury is an environmental pollutant produced by natural phenoms, industrial and mining activity, as well as by deforestation, which is toxic to the human neurodevelopmental period [1]. In the Amazon region, natural sources and anthropogenic actions represent the major causes of mercury exposure, which can be found as organic and non-organic chemical forms, in the riverine populations [2,3,4]. The non-observed adverse effect level (NOAEL), declared by the World Health Organization (WHO), in mercury concentrations of 10 μg g−1 in human hair has been exceeded in contaminated population regions, such as mining areas [5]. Despite the WHO NOAEL recommendation, it fails to delimitate relevant factors as low levels of long-term exposure still occurring or the past acute intoxication still detected in hair [6]. Thus, the establishment of secure levels of mercury exposure is complex, as well as the design of intoxication paradigm in animal models, which could reflect human exposure. Furthermore, mercury distribution, toxicity, and metabolism depend on its chemical form, for which methylmercury (MeHg), an organic derivate, has been extensively studied due to its ability to cross the blood–brain barrier, reaching high levels in the central nervous system (CNS) [7]. Therefore, in vitro and in vivo studies have been essential to elucidate the toxicological mechanisms that underlie the symptoms exhibited by humans following mercury exposure. In experimental challenges, mercury has been administrated by inhalation, oral, or intraperitoneal routes, while in humans the consumption of contaminated foods (i.e., fishes and seafood) reflects the most common intoxication profile. Nonetheless, doses that elicit mercury blood and hair levels similar to those found in clinical studies, in addition to differentiation of acute and chronic exposure, consist of strategies to minimize limitations related to animal studies.
Neurotoxicity of mercury has been described through in vitro and in vivo studies. The occurrence of these undesired neurological effects depends on the level of exposure, by which paresthesia, dysarthria, progressive constriction of the visual field, hearing loss, sensory deficits, ataxia, tremor, deterioration of cognitive functions and paralysis have been reported [8,9,10,11]. Epidemiological and experimental studies have reported the vulnerability of the developing brain to environmental pollutants such as MeHg, the most toxic form of mercury, including a decrease in cell proliferation in the developing neural tube [7,8,9,10]. Several mechanisms have been suggested, including negative effects on neurotransmitter systems, induction of oxidative stress, microtubule disruption, and intracellular calcium homeostasis disturbance [12,13,14,15]. The fundamental toxicological mechanism related to the CNS damage appears to rely on the induction of an excessive amount of synaptic glutamate (i.e., inhibition of reuptake by astrocytes), as well as stimulation of neuronal release of this neurotransmitter, consequently leading to excitotoxicity and cell death [16,17,18]. In addition, mercury also disturbs the cellular redox homeostasis, with overproduction of the reactive oxygen species (ROS) and reduction of antioxidant systems activity [19,20,21,22].
Beyond the detrimental effects of this heavy metal, the concomitant exposure to other neurotoxicants may increase the potential damage on CNS. In this sense, we highlight that the ingestion of ethanol (EtOH), which the intake initiates early in adolescence, has been commonly observed in mining regions, and perhaps may synergically intensify the mercury damage [1]. EtOH directly reduces neuronal plasticity in the early life, impairing cognitive function, emotionality, and motor skills, among other CNS domains in adult life [23,24]. Adolescence is known as a developmental period marked for several changes, including mood, at which point the susceptibility to disorders such as depression, anxiety, and insomnia increases due to hormonal and neuronal functions that undergo an intense process of modifications [25,26].
2. Toxicokinetic Interaction
Robust literature has indicated that mercury exposure deposits in body systems during neurodevelopment with distinct differences among body tissues [1,28,29]. Previous studies already have pointed to such differences in MeHg affinity for CNS regions [30].
Of note, EtOH modify MeHg toxicokinetic in the simultaneous exposure [1,28,31,32]. A survey related to women who drank alcohol concomitantly to a regular consumption of fish/seafood during pregnancy support this interaction [31]. Grandjean and Weihe [31] detected low levels of mercury on the cord-blood of children whose mothers had consumed EtOH compared to those who were abstinent during pregnancy The authors attributed this mercury reduced level to the presence of protector compounds on fish (i.e., polyunsaturated fatty acids), since the elevated frequency of fish consumption in pregnancy has been correlated to a lower average of mercury on cord-blood [31]. However, in a previous study, the authors fail to show that an acute low dose of EtOH exposure (1.0 g/kg) interferes with mercury tissue content [29]. We hypothesize that such disaccord relies on the mercury plus EtOH challenge, which a single dose of both toxicants, dosage, and only 6 h as a cut-off for mercury content evaluation, were not sufficient to observe the scenario of kinetics interactions. In contrast, Tamashiro et al. [32] suggested that animals who intake higher doses of EtOH (5% and 10%) for 10 consecutive days present higher mercury deposits in body tissues, such as the brain, which has been contested by another experimental assay [33].
3. Behavioral Alterations
3.1. Depression
Essentially, the neurochemical basis of MeHg-induced depression-like behavior relies on disturbances in different neurotransmitter systems, mainly glutamate, in the early-exposure, followed by long-lasting changes in brain functioning [39,46,50]. In rats, 30 μM of MeHg modifies glutamatergic synaptic transmission on the hippocampus, through changes on the glutamatergic terminal axon regions, modifying synaptic potential response and resulting in irreversible depression of synaptic efficacy [51]. In the glutamatergic pathway, an increment of glutamate concentrations in the synaptic cleft occurs that results in hyperactivation of N-methyl D-aspartate (NMDA)-type glutamate receptors, leading to an increase of intracellular Na+ and Ca2+ levels, which has been associated with generation of ROS, as well as the triggering of important pathways involved in cell death induced by increased extracellular Ca2+ levels, disrupting glutamate and calcium (Ca2+) homeostasis in intracellular compartments, including mitochondria [7,52,53,54,55,56,57,58,59]. The augment of Ca2+ levels provoke activation of important vias involved in cell death [55]. Such alterations impact on the mitochondrial electron transport chain (ETC), which in vivo and in vitro studies have revealed that MeHg toxicity alters the complexes II and III of the mitochondrial ETC, eliciting depression of respiratory mechanisms and ATP production, and swelling of the mitochondrial matrix, which may contribute to the pathophysiology of depression [56,60]. In fact, alteration in Ca2+ homeostasis; direct toxic effects on mitochondria resulting in mitochondrial damage/dysfunction; and induction of oxidative stress consists of aggravating factor in fundamental brain areas related to mood disorders (i.e., hippocampus) [61], including depression elicited by MeHg exposure [56,60].
In addition, brain-derived neurotrophic factor (BDNF) secreted by astrocytes regulates synaptic plasticity and memory formation in the brain, stimulates release of neurotransmitters in presynaptic neurons, and enhances ion channels in postsynaptic neurons [62,63,64,65]. BDNF also provides neuroprotection on the hippocampal region against ischemic damage, favoring the increase of antioxidant enzymes [3,64,66]. Studies have demonstrated that BDNF undergoes down regulation by MeHg exposure following prenatal, neonatal, and adulthood exposure in rats [67,68,69,70]. Thus, it is reasonable to infer that MeHg predisposes to depression disorders by a plethora of molecular mechanisms. In support of this hypothesis, studies have linked the depression-like behavior associated to the reduction of different Bdnf transcripts levels on the frontal cortex and hippocampus following MeHg exposure [39,69,70].
3.2. Anxiety
Adolescence is a developmental period for which major CNS structural and functional changes occur, particularly pruning and maturation of the prefrontal region that participates in the limbic system, as well as the neuroanatomical circuitry of anxiety [79]. Curiously, the prefrontal cortex and hippocampus that undergo maturation during adolescence also have been described as targets of the toxic effects of mercury, consisting of two fundamental structures for emotional behavior [1,13].
Belém-Filho and colleagues [1] observed increased mercury content levels in the prefrontal region compared to other brain areas associated to anxiogenic-like behavior. Furthermore, reduced GABAergic signaling sensibility on the prefrontal cortex supports the mercury hazardous effects related to anxiety [80].
In vivo experimental analyses have reported pathophysiological changes on the hippocampus after perinatal and postnatal exposure to MeHg by which the neurotoxic effects at cellular level have been correlated with oxidative stress, excitotoxicity, damage to deoxyribonucleic acid (DNA), neurogenesis alterations, calcium (Ca2+) homeostasis disruption, neuroinflammation, and cell death mechanisms [13].
Furthermore, in cortical cell cultures from animals exposed to MeHg during pregnancy, basal levels of extracellular glutamate were higher, while levels of extracellular glutamate evoked-KCl were lower than control sample [82].
In addition, the hypothalamic–pituitary–adrenal (HPA) axis function has been affected by MeHg, which may result in anxiety disorders [83]. In other words, anxiety induced by MeHg during the neurodevelopmental stages shares some molecular pathways observed in depression pathophysiological mechanisms described anteriorly.
3.3. Insomnia
Although insomnia has not been considered an emotional disorder, this disturbance commonly integrates the symptomatology of depression, anxiety, and other psychiatry disorders. Costa Junior et al. [27] conducted epidemiological research focused on emotional and motor manifestations in Amazonian riverine populations exposed to mercury. Residents from Itaituba City that presented higher levels of mercury than those of the Acará City population, have suffered insomnia isolated or associated to emotional disturbances. Posteriorly, Arrifano et al. [5] has accessed CNS disorders in riverine communities chronically exposed to MeHg. In 41.9% of patients presented with high levels (≥10 μg g−1) and 47.7% low levels (≤10 μg g−1) of mercury in hair and exhibited insomnia. In this research, alcohol drinking consisted of an exclusion criterion, which permits to deduce that this response is not related to EtOH intake. Although these two studies have not detected a direct correlation, such symptoms were recorded, highlighting the importance of identifying sleep biomarkers in MeHg intoxication.
Sleep cycle is regulated by environmental conditions (i.e., luminosity), hormonal signals, and neurotransmitter release. Melatonin is the main hormone reported as regulator of dark/light cycle, whereas other biomarkers, as cortisol, can also influence the initiation and duration of the sleep process [92]. Neurotransmitters also have been directly involved in induction and transition phases of sleep, in which the adequate function of all neurotransmitter systems, as well as other dependent processes may impact on cognition, general organic recovery, emotionality, etc. [92]. In a canonical pathway, reduction of luminosity, levels of substances on blood and brain (glucose, cortisol, adrenaline, among others), release of melatonin, followed by increases in GABAergic and galanin transmission results in the end of the wake phase [92,93,94]. On the other hand, imbalance on activity of other regulators such as serotonin, noradrenalin, dopamine, histamine, and glutamate also may result in sleep disturbance [94,95].
In fact, there are two process of sleep regulation called homeostatic and circadian, in which the former regulates the sleep length and depth, whereas the later regulates patterns of maximum sleepiness and maximum alertness throughout the 24 h day [96,97].
Some studies have claimed that such altered homeostatic sleep system induces mood disorders (increase of emotional reactivity/impulsivity), decreasing the connectivity between the prefrontal cortex and amygdala, and consequently increasing sleep deprivation, affecting numerous executive functions such as attention, working memory, inhibition, and cognitive flexibility [98].
Possible damage of the HPA axis or stress situation reinforces the idea of sleep disorders that result in release of these hormones, as observed in studies of single administration of MeHg in rats. One hour following subcutaneous 12 mg/kg of MeHg, elevated ACTH levels with reversible peak in the serum have been observed [112]. Moreover, different forms of mercury compounds can deposit on the adrenal gland in the cortex and medulla regions, interfering with adrenal maturation and increasing corticosterone release, affecting HPA axis [113].
In turn, EtOH intake seems to inhibit melatonin secretion dose-dependently, probably by diminution of tryptophan hydroxylase activity and phase delay in arylalkylamine N-acetyltransferase gene expression, beyond impairment in signalization of noradrenaline in the pineal glandule, with reduction of mRNA expression of β1 and α1 adrenergic receptors [114,115]. Additionally, EtOH exposure induces neurological adaptations, especially in conditions of alcoholism, in which up-regulation in glutamatergic function and down-regulation in GABA signaling result in symptoms related to agitation and increased alertness [116,117].
In the HPA axis, EtOH elicits HPA axis disturbances, mainly during withdrawal syndrome. Primarily, EtOH alters CRH releasing, impairing ACTH levels [117,118]. Indeed, EtOH induces endocrine modifications in the hormonal secretion in the adrenal gland with increases of cortisol levels [118,119]. Such toxicological mechanisms directly alter the stimulation of the pituitary and glucocorticoid secretion, as well as indirectly-by its metabolite acetaldehyde-, in an ACTH-independent manner, acting in adrenocortical cells [120]. Studies in humans have postulated that negative effects after EtOH heavy drinking paradigm were measured by diurnal changes in plasma cortisol, resulting in high concentrations during acute withdrawal [121]. Together, these present clinical implications in the sleep cycle of circadian rhythms. Mathematical hypothesis reinforces that acute/chronic EtOH exposure augments cortisol levels and amplitude of ultradian cortisol rhythm dose-dependently, consequently impairing HPA axis homeostasis [122].
Thus, considering the mechanisms on HPA axis and endocrine alterations, the association between mercury and EtOH may potentiate the risk of insomnia development. As mentioned, mercury and EtOH exposure increases glutamate activity, in which such shared toxicological mechanism can aggravate this condition. Until the present moment, data about the relationship between both toxicants during neurodevelopment remains unknown, reinforcing the urgent necessity of investigations in this field.
All hypothetical pathophysiological mechanisms related to MeHg and/or EtOH exposure on depression, anxiety, and insomnia are showed in Figure 1.
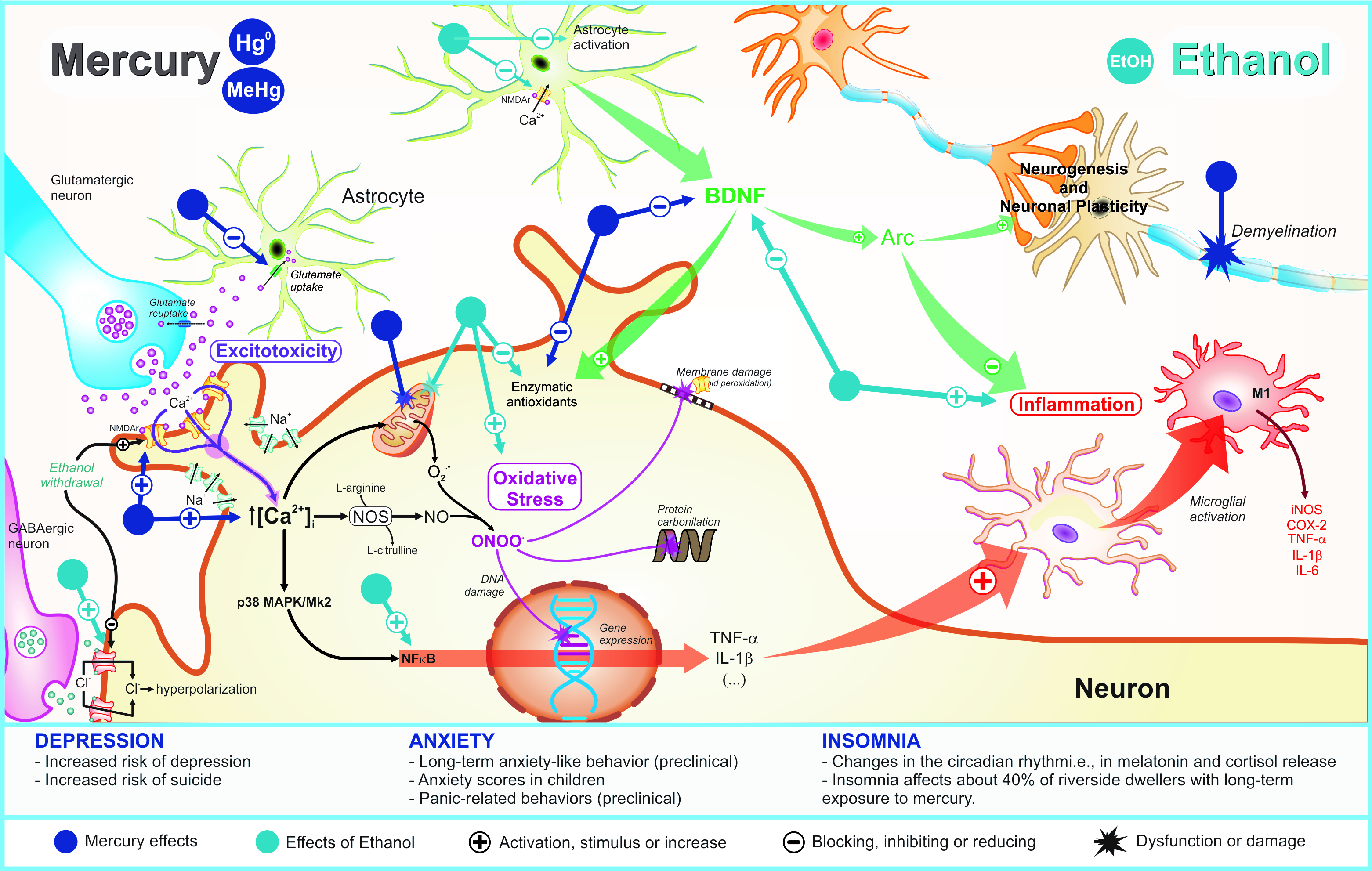
Figure 1. Pathophysiological mechanisms related to deleterious effects of mercury exposure on sleep and emotionality (depression and anxiety) disturbances and its potential synergism with ethanol consumption.
This entry is adapted from the peer-reviewed paper 10.3390/ijms222313131
This entry is offline, you can click here to edit this entry!