Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Ischemia and reperfusion injury comprise complex mechanisms involving disarrangement of the splanchnic microcirculatory flow and impairment of the mitochondrial respiratory chain due to initial hypoxemia and subsequent oxidative stress during the reperfusion phase.
- ischemia-reperfusion injury
- molecular probes
1. Free Radical Synthesis and the Pathophysiology of Ischemia/Reperfusion
When (partial or total) occlusion of the superior mesenteric artery or its branches occurs, splanchnic perfusion is limited due to reduction or, more frequently, interruption of blood flow. Blockage of oxygen supply and an impediment to aerobic energy metabolism induce an acute pathophysiological changes in the affected tissue(s) [23]. The lack of oxygen supply causes tissue ischemia and, if not restored promptly, will result in cellular dysfunction and cell death, ultimately resulting in parietal necrosis [24,25] (Figure 1).
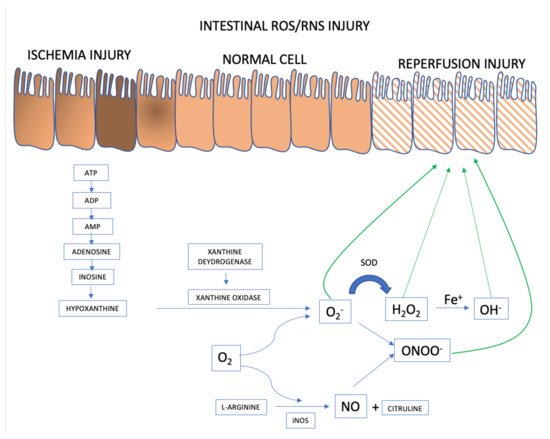
Figure 1. ROS and RNS formation mechanisms in the AMI setting. Adapted from [23].
2. Intestinal Epithelial Cells
ROS and RNS formation begins in the intestinal ischemia phase with adenosine triphosphate (ATP) accumulation generated in the anaerobic metabolism. There is degradation until the accumulation of hypoxanthine that at the beginning of the reperfusion phase, when there is reintroduction of oxygen to the intestinal tissue, interact with xanthine oxidase forming the superoxide anion (O2−), the first ROS formed. From there, the organism launches defenses such as superoxide dismutase (SOD), attenuating and forming ROS such as hydrogen peroxide (H2O2). However, if the response to reperfusion injury continues, the hydrogen peroxide is transformed into hydroxyl from the metal Iron (Fe+) into hydroxyl (OH−), in the so-called Fenton reaction. And in parallel, there may be the activation of RNS with the formation of nitric oxide (NO) from L-arginine, mediated by inducible nitric oxide synthase (iNOS). The combination of NO and superoxide anion forms the highly reactive species called nitrite peroxide (ONOO) which will further damage the intestinal cell’s epithelium.
During the ischemic phase, mitochondrial oxidative phosphorylation is inhibited rendering a drop in the production and storage of adenosine triphosphate (ATP). ATP is successively degraded to adenosine diphosphate (ADP), adenosine monophosphate (AMP), adenosine, inosine, and finally hypoxanthine. Lack of cellular energy causes sodium-potassium (Na+/K+) pump failure resulting in intracellular Na+ accumulation and K+ out of cells, ultimately leading to cellular edema and organelle dysfunction. In addition, an influx of calcium (Ca2+) and chloride (Cl−) ions into the intracellular environment occurs and triggers the activation of calpain protease, which in turn promotes the breakdown of a peptide bridge of the enzyme xanthine dehydrogenase (XDH) and subsequent formation of XO.
Although essential for the rescue of morphofunctional integrity of the affected tissues, restoration of mesenteric blood flow and consequent ischemic tissue reoxygenation has a deleterious effect because, paradoxically, reperfusion itself aggravates the damage [26,27]. Oxygen together with hypoxanthine and XO, synthesized during ischemia, catalyze the formation of ROS [28,29]. Re-introducing oxygen into the visceral circulation via reperfusion leads to the formation of O2− and hydrogen peroxide (H2O2) after successive monovalent reductions. In the presence of iron, copper, cobalt, chromium, or vanadium, the production of highly reactive hydroxyl radical (OH.) is promoted via the Haber-Weiss and Fenton reactions [30]. There is an activity burst of the oxidative process characterized by the abundant production of multiple ROS and RNS within a few minutes after the restoration of blood flow [27]. The events underlying the damage caused by ischemia/reperfusion produce an uncontrolled and excessive release of ROS and RNS that overcome the organic line of defense represented by free radical scavengers [31].
The mitochondrial respiratory electron transport chain is the main intracellular site of ROS production and polymorphonuclear leukocytes play an important role in several pathological conditions also generating free radicals and nitric oxide (NO) synthesis. Different forms of mitochondrial dysfunction and tissue inflammation can affect the organ undergoing ischemia and reperfusion and may even compromise other organs and systems with a paracrine or and endocrine effect. This phase can lead to the failure of multiple organs and systems [23,32].
Nitric oxide (NO) dynamics underpin changes involving RNS. NO is produced from L-arginine by three main isoforms of nitric oxide synthase (NOS): epithelial NOS (eNOS), related to vasodilation and vascular regulation; neuronal NOS (nNOS), linked to various intracellular signaling pathways; and inducible NOS (iNOS), which has been reported to have beneficial microbicidal, antiviral, antiparasitic and antitumoral actions, but has also been implicated in the pathophysiology of colitis [33]. While the production of NO by nNOS and eNOS is regulated by a Ca2+/calmodulin-dependent mechanism, iNOS is activated in response to triggers such as endotoxins or cytokines, which can lead to rapid production of large amounts of NO. Several diseases have been associated with excessive levels of NO production, resulting in serious deleterious cell-physiological consequences [34,35,36,37,38]. All products formed by NO reactions are collectively called RNS. Despite the discovery of NO as an endothelium-derived relaxing factor, it plays a critical role in the pathophysiology of sepsis as an important mediator of endotoxin-induced arteriolar vasodilatation, hypotension, and shock [39]. At high concentrations, NO is importantly involved in inflammatory, infectious, and degenerative diseases [40]. Via reactions with other free radicals produced during oxidative stress, NO can be converted to nitrogen dioxide (NO2), peroxynitrite (ONOO−), and dinitrogen trioxide (N2O3). NO2 is formed from NO autoxidation (reaction of NO with oxygen). ONOO− is a powerful electron oxidant and is formed through the diffusion-controlled reaction between O2− and NO; its most relevant targets are peroxiredoxins, glutathione peroxidase (GSH), CO2, and metal centers. N2O3 can be formed from a reaction between NO2 and NO and is considered an important intermediate in the autoxidation of NO. N2O3 is rapidly hydrolyzed to NO2 [41]. All these compounds can subsequently react with various classes of biomolecules, including lipids, DNA, thiols, amino acids, and metals, leading to oxidation and nitration. If produced at high levels, RNS will detrimentally impact cell function, leading to changes in membrane integrity, loss of enzyme function, and DNA mutations [42].
It is noteworthy that, despite its typically beneficial antioxidant and vasodilatory functions, NO in high concentrations induces caspase-mediated apoptosis of epithelial cells in the intestinal tissue during ischemia and reperfusion. In addition, O2− rapidly reacts with NO to produce ONOO−, which is another potent oxidant [43]. In the vasculature, the reaction of NO with O2− leads to the formation of ONOO− and decreases the vasorelaxant efficacy of NO. ONOO− is a strong oxidant that can hydroxylate aromatic amino acids, oxidize thiols and lipids, and nitrate-free and protein-bound tyrosine residues. The number of possible reactions leading to secondary RNS formation illustrates the strong potential of NO to contribute to oxidative damage. High concentrations of NO, particularly in combination with increased oxidant production, cause tissue damage and inflammation through the production of NO2, ONOO− and other nitrating, nitrosating, and oxidizing intermediates, and via inhibition of metal-dependent enzymes [44,45].
Several enzymes, such as cytochrome P450, the enzyme complexes of the mitochondrial respiratory chain, XO [46], eNOS [47], heme oxygenase (HO) [48], myeloperoxidase (MPO) [49], lipoxygenase (LOX), cyclooxygenase (COX) [50], and NADPH oxidases (NOX) [51] generate ROS under pathological conditions leading to oxidative stress [52]. All these factors contribute to persistent oxidative stress in the cellular environment, which will result in progressive functional impairment of critical intracellular organelles and structures, including membranes, mitochondria, the endoplasmic reticulum, the cytoskeleton, and the nucleus. These deleterious effects occur mainly due to the oxidation of proteins, DNA, and lipids, ultimately culminating in cell death [53,54].
A balance between ROS levels and the activity of inactivating (antioxidant) enzymes is crucial for the maintenance of cellular homeostasis. Erythroid-related nuclear factor 2 (Nrf2) is a transcription factor that plays an important role in the response to oxidative stress to maintain redox balance. Under homeostatic conditions, Nrf2 is bound to its chaperone Keap1 (Kelch-like ECH association protein 1) in the cytoplasm. However, when oxidative stress occurs, Nrf2 dissociates from the inactive Keap1-Nrf2 complex and translocates to the nucleus, where it regulates specific gene expression to induce the synthesis of antioxidant enzymes [55]. O2− and H2O2 are inactivated by superoxide dismutase and catalase or the glutathione peroxidase system, respectively. OH. is typically more harmful than these ROS, as this oxygen-derived free radical does not have an intracellular inactivator. Its production intensifies the severity of injuries to cell structures, causing DNA damage caused by adducts of lipid peroxidation, and the production of other free radicals (such as malondialdehyde, hydroperoxide, and ONOO−, among other substances capable of stimulating the adherence of granulocytes to the microvascular endothelium [55,56].
3. Molecular Probe Fundamentals
Oxidative and nitrosative stress biomarkers are important tools to assess the balance between reactive species and antioxidants, contributing to the understanding of the pathophysiology of diseases [57]. Direct measurement of your cellular levels is a challenge, as direct and accurate measurement is complex, due to its short productive life and fast reactivity with other REDOX regulators [58]. Fluorescent probes for ROS selectively assess cellular levels of ROS in a very simple way, but it is important to consider their limitations. Fluorescent probes are able to monitor the behavior of a target biomolecule in live cells in real time [59].
The dihydrorhodamine 123 (DHR123) probe passively diffuses the cell membrane and concentrates in the intracellular space. In the presence of H2O2, hypochlorous acid (HOCl), or ONOO−, it is oxidized to rhodamine (R123) which exhibits green fluorescence. DHR123 is considered an intracellular probe for general detection of ROS; however, it has a lower stability than several other commercially available probes.
The CM-H2DCFDA (5-diacetate and 6-chloromethyl-2′,7′-dichlorodihydro-fluorescein) probe passively crosses the plasma membrane to enter the cell after which its acetate groups are cleaved by esterases to generate intracellular CM-H2DCF; the thiol-reactive chloromethyl group reacts with intracellular glutathione and other thiols, and subsequent oxidation renders a fluorescent intracellular adduct. This probe is used to detect intracellular ROS and can react with H2O2, OH, ONOO− and other peroxide radicals. However, it is easily auto-oxidized resulting in a spontaneous increase in fluorescence, which must be corrected for at the time of the reading, discounting the value of a cell-free well containing the probe, as described by Hempel et al. Although this type of probe mainly detects H2O2, OH, and ONOO−, it is not specific for any oxidant because it responds to a wide range of oxidizing reactions; the CM-H2DCFDA probe is therefore considered a probe for general detection of ROS [60].
Fluorogenic complex probes containing boronate are used as a basis for detecting intracellular H2O2. Aromatic boronates react with H2O2, to generate a corresponding phenol, forming a highly fluorescent molecule in cells. Arylboronates also react with ONOO−, six times faster than with H2O2, verified by flow kinetics technique and high performance liquid chromatography (HPLC) analysis [61]. One of the characteristics is its photophysical properties, such as high photostability and suitable high fluorescence. In addition, the iminocoumarin by-products have excitation and emission wavelengths that are longer, whereas rapid cyclization would generate the highly fluorescent benzothiazolyl iminocoumarin [62].
Amplex Red reagent is a colorless, highly sensitive, non-fluorescent compound used as a stable probe to detect the generation of H2O2. It is oxidized by horseradish peroxidase (HRP) to a fluorescent product, resorufin. One of the main complicating factors is photochemical oxidation in the presence of biological reducers (glutathione) that induce the formation of free radicals (O2− and H2O2), making the measurement of intracellular H2O2 a problem, even in the absence of HRP and H2O2. It is a highly sensitive method for detecting H2O2 and resorufin is stable for some time. However, it is impervious to cells and cannot be used to detect intracellular H2O2. Amplex Red is a very sensitive method for detecting ROS in organelles, as well as extracellular ROS, which is freely diffusible. The Amplex Red assay is also used to evaluate ROS formation in mitochondria [61,63].
CellRox represents another class of probes used for the general detection of ROS and comes in different models capable of emitting distinct fluorescence signals. In a reduced state, these cell-permeant dyes are non- or weakly fluorescent and become fluorescent upon oxidation by ROS. In general, CellRox can be oxidized by OH. and O2−, while CellRox orange is also capable of detecting H2O2, NO and ONOO-. These probes exhibit outstanding photostability compared to DCF [64] and it has also been shown that these probes can detect signals not detected by DCF [65]. Furthermore, depending on the model, can be used for in situ detection, allowing the assessment of real-time ROS dynamics in any given tissue [66].
The dihydroethidium (DHE) probe is capable of specifically detecting O2− radicals in intracellular and extracellular environment [67]. In addition, it can also be used to detect O2− in situ. The primary radical hydroethidine is derived from the loss of an aromatic amino hydrogen atom that, upon rearrangement, further reacts with another O2− anion to form DHE. Acetylation of the aromatic amino groups in hydroethidine inhibited its reaction with O2− [68]. MitoSox is the preferred probe for the specific analysis of mitochondrial O2−; this reagent selectively targets mitochondria where it is rapidly oxidized by O2− (but not by other ROS or RNS) producing a red fluorescent signal, the oxidized product is highly fluorescent upon binding to nucleic acid [69].
DAF-FM (4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate) is the leading molecular probe for the detection of NO. Like CM-H2DCFDA, DAF-FM diacetate also passively diffuses the plasma membrane and is cleaved by esterases to generate intracellular DAF-FM. Subsequent oxidation by NO yields a triazole product accompanied by increased fluorescent recovery [70]. DAF-FM is not a reversible balance sensor, which limits its ability to track rapid target substance (NO) fluctuations in real time.
The aminophenyl fluorescein (APF) and hydroxyphenyl fluorescein (HPF) probes provide better selectivity and stability than CM-H2DCFDA for specific detection of OH. and ONOO− with relatively high resistance to light-induced oxidation. In their initial (reduced) form, the APF and HPF molecular probes are not fluorescent until they react with ONOO− or OH, producing bright green fluorescence [45], resulting in cleavage of the aminophenyl ring from the fluorescein ring system, which is highly fluorescent. APF will also be transformed into the fluorescent form if exposed to a combination of H2O2 and horseradish peroxidase (HRP); HRP catalyzes the oxidation of APF by H2O2 [71].
This entry is adapted from the peer-reviewed paper 10.3390/metabo11120802
This entry is offline, you can click here to edit this entry!