The rapid mutation of the SARS-CoV-2 virus is now a major concern with no effective drugs and treatments. The severity of the disease is linked to the induction of cytokine storm that promotes extensive inflammation in the lung, leading to many acute lung injuries, pulmonary edema, and eventually death. Mesenchymal stem cells (MSCs) might prove to be a treatment option as it has immunomodulation and regenerative properties. Clinical trials utilizing MSCs in treating acute lung injury (ALI) or acute respiratory distress syndrome (ARDS) has provided a basis in treating post-COVID-19 patients.
1. Background
The rapid mutation of the SARS-CoV-2 virus is now a major concern with no effective drugs and treatments. The severity of the disease is linked to the induction of a cytokine storm that promotes extensive inflammation in the lung, leading to many acute lung injuries, pulmonary edema, and eventually death. Mesenchymal stem cells (MSCs) might prove to be a treatment option as they have immunomodulation and regenerative properties. Clinical trials utilizing MSCs in treating acute lung injury (ALI) or acute respiratory distress syndrome (ARDS) have provided a basis in treating post-COVID-19 patients.
2. Immunomodulatory Effects of MSCs in COVID-19
Mesenchymal stem cells (MSCs) are well known for their promising cell-based therapies in infectious diseases due to their immunomodulatory potentials in managing inflammatory diseases
[1][2]. According to Weiss and Dahlke
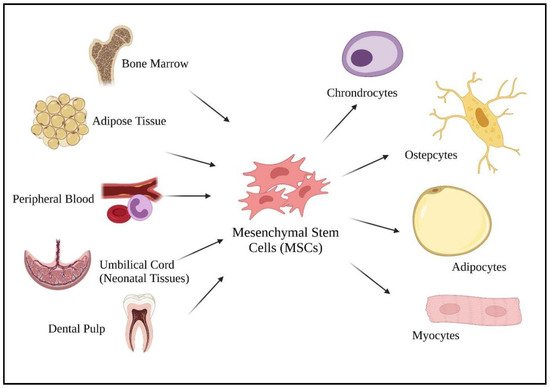
[3], MSCs can be harvested from various adult tissues, from bone marrow to adipose tissue, dental pulp, peripheral blood, as well as from neonatal tissues (umbilical cord) (
Figure 1). Compared to fetal and umbilical stem cells, adult stem cells are preferable as there are fewer ethical issues, widespread availability, and are high in clinical applications. As a multipotent cell, MSCs give rise to other descendent lineages such as chondrocytes, osteocytes, adipocytes, and myocytes (
Figure 1). The ability to differentiate into various descendant cell lineages allows the use of stem cell therapies in treating chronic diseases, such as COVID-19.
Figure 1. Sources of mesenchymal stem cells and their differentiated lineage. Created with
BioRender.com.
2.1. Immunological Mechanism during COVID-19
As mentioned previously, the best defense against COVID-19 is the immune system, as it sets up the natural ability of the body to defend against pathogens. COVID-19 will not affect us as long as our body has good immunity
[4]. However, when an individual is infected by COVID-19, the virus will induce cytokine secretion and localize inflammation
[5]. The over-response of the immune system leads to the release of cytokines, chemokines, and other immune effector cells that are proinflammatory, causing cell damage. In this situation, MSCs will exhibit their immunosuppression properties to suppress the overreaction of the immune system towards COVID-19. Kavianpour et al.
[5] also stated that the MSCs will locate at the inflamed lung tissues and secrete factors that can modulate the immune system. This will enable the prevention of oxidative stress (ROS) and fibrosis of the lung tissue.
In dealing with COVID-19, both innate and adaptive immunity are involved. According to WHO
[6], the immunological mechanism of COVID-19 infection involves two phases. The first phase is the activation of innate immunity. Similar to other infectious diseases, the first and second lines of defense will restrict the entry of the SARS-CoV-2 virus into the host cells. The immune response will secrete interferon and chemical substances such as cytokines. Interferon will interfere with the replication of viruses in the cells while cytokines cause inflammatory reactions. Specifically, the innate immune response towards the infection mainly depends on the interferon type I protein with its downstream signaling. The viral RNA of the virus will act as the pathogen-associated molecular molecules (PAMPs) recognize by the toll-like RNA receptors (TLR-3 and TLR-7) and cytosolic RNA sensor (RIG-I/MDA5)
[7]. NF-κB and interferon regulatory transcription factor-3 (IRF-3) will be activated and translocated into the nucleus. Inside the nucleus, the expression of IFN type I and proinflammatory cytokines is induced, constructed as the first-line defense against virus entry.
Generally, the infection stops at the first line of defense if the individual immunity is strong enough, or if the viral load is low. This IFN type I response is vital for viral suppression. However, this response is not effective in dealing with SARS-CoV-2 as the response is suppressed, and the virus will induce their programmed cell death, apoptosis. Without it, the virus continues replicating and causes delayed IFN type I response, heralding the influx of macrophages and neutrophils as a source of inflammation and Th1 immune response
[7].
When the virus encounters a weaker immune system, clearance does not occur, resulting in the stimulation of adaptive immunity. Adaptive immunity via T cells and B cells would then provide the protection required. T cells are regulated by the antigen-presenting cells (APCs) by engendering the cytokine environment. It recognizes cells infected with the virus and rapidly differentiates and proliferates. CD8+ cytotoxic T cells clear virus-infected cells leading to reduced viral load. CD4+ helper T cells produce proinflammatory cytokines and mediators to further strengthen the response, including stimulating B-cells. B-cell produces antibodies against the virus leading to high amounts of neutralizing antibodies and Th2 cytokines (IL-4, IL-5 and IL-10).
2.2. Immunomodulation Mechanism of MSCs Involving Molecular Signaling
The immunomodulation ability of the MSCs can be exerted in various ways: soluble factors (paracrine interaction), cell–cell contact, and extracellular vesicles (EVs)
[8][9]. The effect of the mechanism is exhibited either individually or in combination with various biomolecules by MSCs on the immune cells such as macrophages and neutrophils, DCs, T cells, B cells, and NK cells (
Table 1)
[1][10].
Table 1. Immunomodulation mechanisms of MSCs with various immune cells.
| Immune Cells |
Mechanism of Actions |
Implicated Biomolecules |
Action Pathway |
Results |
| Macrophages and Neutrophils |
Soluble Factors |
PGE 2 |
Activation of signal transducer activators of transcription-3 (STAT 3) |
M2 macrophage phenotype switch |
| DCs |
Soluble Factors |
PGE 2 |
Lower the expression of CD38, CD80, CD86, IL-12, and IL-6 |
Inhibit DCs maturation |
| CCR7–CCL21 interaction |
Lowering the migratory ability of DCs |
| TNF-α-stimulating gene 6 |
Inactivation of mitogen activated protein kinase (MAPK) and nuclear factor-kappa B (NF-κB) |
Suppress DCs maturation |
| HLA-G |
Blocking the secretion of cytokines such as TNF-α, ΙL1-α, β, IL-6, IL-7, IL-8, IL-9, GM-CSF, and IFN-γ |
Prevent the differentiation of monocytes to DCs |
| EVs |
miR-21-5p, miR-142-3p, miR-223-3p, and miR-126-3p |
Interaction with JAG1, PDCD4, IL-12p35,
downregulation of IL-6 expression |
Inhibition of DCs maturation |
| T Cells and B Cells |
Soluble Factors |
PGE2 |
CAMP production in T cells |
Downregulate the IL-2 and IL-2R expression |
| Negatively regulating the phosphatidylinositol hydrolysis and the diacylglycerol and inositol phosphate (IP) production |
T cells inactivation |
| Orchestrating regulatory T (Treg) responses |
Promote Th2 immune response |
| IDO |
Blocks the metabolism from tryptophan to kynurenine in combination with TGF-β1 and HGF |
Suppress T cells proliferation |
| NO |
Activating the transcription 5 phosphorylation |
Inhibition of TCR-mediated T cells proliferation and inflammatory cytokine production |
| Galectin 1 and 3 |
Preventing the clustering of TCR via crosslink interaction mechanism |
Suppress the proliferation of T cells proliferation |
| HLA-G with IDO and IL-10 |
Suppress proliferation of T cells |
Indirectly blocked the secretion of cytokine (TNF-α, ΙL1-α, ΙL1-β, IL-6, IL-7, IL-8, IL-9, GM-CSF,
and IFN-γ) |
| Cell–Cell Interactions |
Fas/Fas ligand death signaling pathway |
Downstream activation of the Fas-associated death domain and caspases |
T cells apoptosis |
| TNF-related apoptosis-inducing ligand (TRAIL)/death receptor (DR) signaling pathway |
High production of TRAIL and binds to DR on T cells |
T cells apoptosis |
| Programmed death ligand-1 (PD-L1)/programmed death-1 (PD-1) |
Inhibition of MAPK followed by Src-homology 2 domain containing protein tyrosine phosphatases (SHP)-1 and SHP-2 phosphorylation |
Reduces T cells proliferation |
| NK Cells |
Cell-cell Interaction |
HLA class I |
Upregulate the expression of HLA class I molecules to interact with killer cell immunoglobulin-like receptors (KIRs) |
Inhibit cytolytic activity of NK cells |
| Interacting with KIR2DL4 |
Inhibit NK cells and cytokine production |
| Toll-like receptors (TLRs) |
Activation of TLR3 |
Increases the immunosuppression against NK cells |
| Soluble Factors |
IDO and PGE2 with aid from TGF-β1 and HGF molecules |
Inhibit IL-2 induced NK response |
Immunosuppression of NK cells |
2.2.1. Immunomodulating Effects of MSCs on Macrophages and Neutrophils
Based on a study by Mallis et al.
[11], MSCs can modulate the macrophage phenotype (M1/M2) via cell–cell or paracrine interaction. Macrophages and neutrophils are important in the antigen presentation process to dendritic cells (DCs) that will stimulate cellular immunity. M1 phenotype macrophages, which are classically activated, are responsible for phagocytosis of pathogens, and present their antigen epitopes to DCs. This process will stimulate the release of cytokines (TNF-α, ΙL-1β, ΙL-1α, IL-6, IL-12) that cause inflammation by M1 macrophages and activate the Th1 immune response. Alternately, M2 macrophages (alternatively activated) promote the Th2 immune response. Th2 immune response counteracts with Th1 immune response, having immunosuppressive properties. These cells are high in anti-inflammatory molecules, associated with tissue repairs and cell apoptosis clearance. This cell–cell interaction enables MSCs modulation of macrophage phenotypes.
The presence of IFN-γ activates MSCs and results in the production of TNF-α, MCP1, and IL-1β. These secreted soluble factors can advance the phenotype of M1 macrophages. On the other hand, MSCs also express prostaglandins E2 (PGE2), which will induce an M2 macrophage phenotype switch. A study has shown that the activation of signal transducer transcription-3 (STAT3) occurs via cell–cell interactions between MCSs and the macrophages
[12]. STAT3 transcription factors are responsible for IL-10 production that promote immunosuppressive functions.
2.2.2. Immunomodulating Effects of MSCs on DCs
MSCs can interfere with the maturation of DCs via the production of soluble factors. Factors such as TNF-α, IL-1β, and IL-6 produced by M1 macrophages and IFN-γ will activate the MSCs and drive the maturation of DCs. Alternatively, PGE2 secreted by the activated MSCs is vital in the inhibition of DCs maturation. This soluble factor is crucial in preventing further damage to the lung cells and tissues. According to a study by Liu et al. and Sadeghi et al.
[9][13], inhibition of DCs maturation by PGE2 interfere with T cells responses, as there are lower levels of CD38, CD80, CD86, IL-12, and IL-6 which are important for T cells activation and lead to cytokine storm. PGE 2 also lowers the migratory ability of DCs via CCR7–CCL21 interaction. Moreover, Liu et al.
[13] also suggested that a stimulating gene produced by TNF-α produced by MSCs can also suppress DCs maturation by inactivating the signaling cascades mediated by mitogen-activated protein kinase (MAPK) and nuclear factor-kappa B (NF-κB). HLA-G soluble factors also prevent the differentiation of monocytes to DCs by blocking the secretion of cytokines such as TNF-α, ΙL1-α, β, IL-6, IL-7, IL-8, IL-9, GM-CSF, and IFN-γ. Furthermore, extracellular vesicles (EVs) that contain specific miRNAs such as miR-21-5p, miR-142-3p, miR-223-3p, and miR-126-3p also contribute to the inhibitory of maturation of DCs.
2.2.3. Immunomodulating Effects of MSCs on T Cells and B Cells
There are several ways of MSCs immunomodulation via regulation of T cells. For example, secretion of molecules that affect the T cells responses and the cell–cell contact positively and negatively to inhibit the T cells proliferation. The production of PGE2, indoleamine-2,3-dioxygenase (IDO), TGF-β, and hepatocyte growth factor (HGF) will effectively inhibit the proliferation of T cells
[14]. According to Mallis et al.
[11], PGE2 is a prostanoid, responsible for T cells activation by the production of cAMP and can exert immunosuppressive properties on T cells. The cAMP produced can downregulate the IL-2 and IL-2R expression which will contribute to the activation of T cells receptors. PGE2 will also inactivate the T cells by negatively regulating the phosphatidylinositol hydrolysis and the diacylglycerol and inositol phosphate (IP) production. The immunosuppression mechanism of MSCs via T cells also can be shown by the T cells polarizing that will promote the Th2 immune response and orchestrate regulatory T (Treg) responses. Moreover, the immunosuppressive response also happens when blockage of metabolism by IDO from tryptophan to kynurenine is essential for T cell cycles
[14]. When IDO is combined with TGF-β1 and HGF, T cells proliferation will be suppressed. The NO signaling pathway also suppressed the T cells response by activating the transcription 5 phosphorylation, resulting in the inhibition of TCR-mediated T cells proliferation and inflammatory cytokine production
[11][14]. Galectins 1 and 3 also effectively suppress the proliferation of T cells proliferation by preventing the clustering of TCR via a crosslink interaction mechanism. There are also soluble HLA-G factors that can suppress the proliferation of hyperactive T cells with the presence of IDO and IL-10.
For cell–cell interactions, MSCs regulate their immunomodulatory properties by T cell apoptosis. According to Mallis et al.
[11], Fas/Fas ligand death signaling pathway can lead to cell death of T cells. This action happens by the downstream activation of the Fas-associated death domain and caspases. MSCs will express Fas ligands when encountering inflammation stimuli. The Fas ligand binds to the Fas receptors of the hyperactivated T cells. TNF-related apoptosis-inducing ligand (TRAIL)/death receptors (DRs) signaling pathway is another potential activator for the Fas-associated death domain. IFN-γ causes the high production of TRAIL and binds to DRs on T cells, causing apoptosis. Programmed death ligand-1 (PD-L1)/programmed death-1 (PD-1) interaction also reduces T cells proliferation. The inhibition of MAPK due to the binding of PD-L1 to the PD-1 molecules of T cells, followed by Src-homology 2 domain containing protein tyrosine phosphatases (SHP)-1 and SHP-2 phosphorylation inhibits cellular proliferation.
MSCs immunomodulate B cells via secretion of soluble factors and cell-cell interaction. Both of these pathways are similar to the T cell’s regulation response. IDO, PGE2, the production of TGF-β1, and HGF can lead to cell cycle arrest of B cells. Fas/Fas ligand, TRAIL/DR death signaling, and PD-L1/PD-1 pathways promote the apoptosis of B cells. The secretion of GM-CSF by MSCs also inhibits the secretion of CXCR4, CXR5, IL-6, and IL-7 by activated B cells, effectively preventing the migratory ability and homing of B cells towards CXCL12 and CXCL13 chemoattractant agents and resulting in the initiation of inflammation
[11]. MSCs are effective in modulating the B cell’s secreted molecules, even so, it has no negative effects on the IFN-γ, TNF-α, IL-4, and IL-10 expressions by B cells.
2.2.4. Immunomodulating Effects of MSCs on NK Cells
NK cells are responsible for eliminating virus-infected cells from the host. MSCs activated by IFN-γ will upregulate the expression of HLA class I to interact with killer cell immunoglobulin-like receptors (KIRs). In this way, cytolysis mediated by NK cells is inhibited. Furthermore, toll-like receptors (TLRs) presented at MSCs are important as the activation of TLR3 increases the immunosuppression against NK cells. HLA-G will also interact with KIR2DL4 to modulate the function of NK cells
[11]. Last but not least, IDO and PGE2 inhibit NK response with aid from TGF-β1 and HGF molecules.
This entry is adapted from the peer-reviewed paper 10.3390/ijms222212421