Together with histone variants and modifications, alterations in nucleosome positioning, non-coding RNAs, and DNA methylation constitute the epigenetic toolkit. DNA methylation describes the chemical modification of the DNA itself by the addition of methyl groups mostly on cytosines, but also on adenines via DNA methyltransferases (DNMTs) [60], with DNMT1 and DNMT3A being the major DNMTs in the CNS [61]. DNA methylation effects, i.a. transcriptional control when occurring at enhancer and promoter sites, alternative promoter choice and alternative splicing [62,63]. At the level of transcriptional regulation, methylated motifs of transcription factor (TF) binding sites physically impede the binding of methyl-sensitive TFs, leading to transcriptional suppression. Furthermore, the interaction of the methyl-CpG-binding domain proteins (MBDs) with methylated DNA prevents binding of TFs and promotes inactive heterochromatin formation by recruiting other chromatin and nucleosome remodeling factors [64].
- Alzheimer
- tauopathy
- TAU
- MAPT
- epigenetics
- neurodegeneration
- neurogenetic disease
- DNA methylation
1. Introduction
2. Implication of DNA Methylation in AD and Tauopathies
2.1. Age-Dependent Changes of DNA Methylation Marks and the Relevance for AD and Tauopathies
2.2. Evidence for the Implication of Altered DNA Methylation Signatures in AD and Tauopathies
2.2.1. DNA Methylation Changes Lead to Pathological Phosphorylation of TAU
2.2.2. Altered DNA Methylation Signatures as a Consequence of Disease Pathophysiology, Such as Aβ Burden and TAU-Phosphorylation
2.2.3. Aβ Peptide and TAU-Phosphorylation-Driven Changes in the Expression and Localization of DNA Repair Related Proteins
2.2.4. Aβ-Associated Changes in DNA Methylation of Cell Cycle-Related Genes
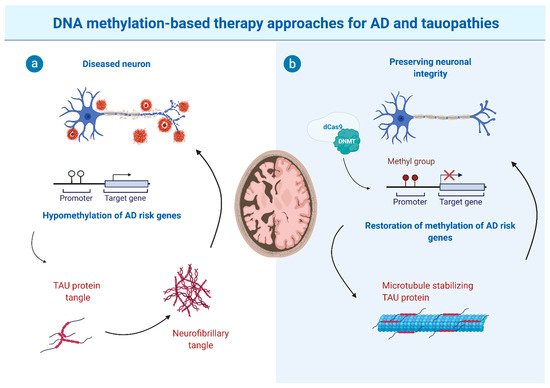
3. The Potential and Limitations of DNA Methylation-Based Therapy Approaches
4. Altered DNA Methylation Signatures as Potential Biomarkers for AD/Tauopathies Disease and Disease Progression?
This entry is adapted from the peer-reviewed paper 10.3390/cells10113064
References
- Zempel, H.; Mandelkow, E. Lost after translation: Missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014, 37, 721–732.
- Zempel, H.; Mandelkow, E. Mechanisms of Axonal Sorting of Tau and Influence of the Axon Initial Segment on Tau Cell Polarity. In Advances in Experimental Medicine and Biology; Springer Nature: Cham, Switzerland, 2019; Volume 1184, pp. 69–77.
- Spillantini, M.G.; Van Swieten, J.C.; Goedert, M. Tau gene mutations in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Neurogenetics 2000, 2, 193–205.
- Barker, W.W.; Luis, C.A.; Kashuba, A.; Luis, M.; Harwood, D.G.; Loewenstein, D.; Waters, C.; Jimison, P.; Shepherd, E.; Sevush, S.; et al. Relative Frequencies of Alzheimer Disease, Lewy Body, Vascular and Frontotemporal Dementia, and Hippocampal Sclerosis in the State of Florida Brain Bank. Alzheimer Dis. Assoc. Disord. 2016, 16, 203–212.
- Höglinger, G.U.; Respondek, G.; Stamelou, M.; Kurz, C.; Josephs, K.A.; Lang, A.E.; Mollenhauer, B.; Müller, U.; Nilsson, C.; Whitwell, J.L.; et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov. Disord. 2017, 32, 853–864.
- Kovacs, G.G. Invited review: Neuropathology of tauopathies: Principles and practice. Neuropathol. Appl. Neurobiol. 2015, 41, 3–23.
- Shi, Y.; Zhang, W.; Yang, Y.; Murzin, A.; Falcon, B.; Kotecha, A.; Van Beers, M.; Tarutani, A.; Kametani, F.; Garringer, H.J.; et al. Structure-based Classification of Tauopathies. bioRxiv 2021, 1–27.
- Dickson, D.W.; Kouri, N.; Murray, M.E.; Josephs, K.A. Neuropathology of frontotemporal lobar degeneration-Tau (FTLD-Tau). J. Mol. Neurosci. 2011, 45, 384–389.
- Spiegel, A.M.; Sewal, A.S.; Rapp, P.R. Epigenetic contributions to cognitive aging: Disentangling mindspan and lifespan. Learn. Mem. 2014, 21, 569–574.
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194.
- Rowbotham, D.A. Epigenetic Changes in Aging and Age-related Disease. J. Aging Sci. 2015, 3.
- Grant, W.B.; Campbell, A.; Itzhaki, R.F.; Savory, J. The significance of environmental factors in the etiology of Alzheimer’s disease. J. Alzheimer’s Dis. 2002, 4, 179–189.
- Bishop, N.A.; Lu, T.; Yankner, B.A. Neural mechanisms of ageing and cognitive decline. Nature 2010, 464, 529–535.
- Lardenoije, R.; Iatrou, A.; Kenis, G.; Kompotis, K.; Steinbusch, H.W.M.; Mastroeni, D.; Coleman, P.; Lemere, C.A.; Hof, P.R.; van den Hove, D.L.A.; et al. The epigenetics of aging and neurodegeneration. Prog. Neurobiol. 2015, 131, 21–64.
- Levenson, J.M.; Roth, T.L.; Lubin, F.D.; Miller, C.A.; Huang, I.C.; Desai, P.; Malone, L.M.; Sweatt, J.D. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem. 2006, 281, 15763–15773.
- Morris, M.J.; Monteggia, L.M. Role of DNA methylation and the DNA methyltransferases in learning and memory. Dialogues Clin. Neurosci. 2014, 16, 359–371.
- Laplant, Q.; Vialou, V.; Covington, H.E.; Dumitriu, D.; Feng, J.; Warren, B.L.; Maze, I.; Dietz, D.M.; Watts, E.L.; Iñiguez, S.D.; et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat. Neurosci. 2010, 13, 1137–1143.
- Xu, S.; Wilf, R.; Menon, T.; Panikker, P.; Sarthi, J.; Elefant, F. Epigenetic control of learning and memory in drosophila by Tip60 HAT action. Genetics 2014, 198, 1571–1586.
- Guan, J.S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.H.; Joseph, N.; Gao, J.; Nieland, T.J.F.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55–60.
- McQuown, S.C.; Barrett, R.M.; Matheos, D.P.; Post, R.J.; Rogge, G.A.; Alenghat, T.; Mullican, S.E.; Jones, S.; Rusche, J.R.; Lazar, M.A.; et al. HDAC3 is a critical negative regulator of long-term memory formation. J. Neurosci. 2011, 31, 764–774.
- Cui, D.; Xu, X. Dna methyltransferases, dna methylation, and age-associated cognitive function. Int. J. Mol. Sci. 2018, 19, 1315.
- Oliveira, A.M.M.; Hemstedt, T.J.; Bading, H. Rescue of aging-associated decline in Dnmt3a2 expression restores cognitive abilities. Nat. Neurosci. 2012, 15, 1111–1113.
- Johnson, A.A.; Akman, K.; Calimport, S.R.G.; Wuttke, D.; Stolzing, A.; De Magalhães, J.P. The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuvenation Res. 2012, 15, 483–494.
- Hahn, A.; Pensold, D.; Bayer, C.; Tittelmeier, J.; González-Bermúdez, L.; Marx-Blümel, L.; Linde, J.; Groß, J.; Salinas-Riester, G.; Lingner, T.; et al. DNA Methyltransferase 1 (DNMT1) Function Is Implicated in the Age-Related Loss of Cortical Interneurons. Front. Cell Dev. Biol. 2020, 8.
- McKinney, B.C.; Lin, C.W.; Rahman, T.; Oh, H.; Lewis, D.A.; Tseng, G.; Sibille, E. DNA methylation in the human frontal cortex reveals a putative mechanism for age-by-disease interactions. Transl. Psychiatry 2019, 9.
- Salameh, Y.; Bejaoui, Y.; El Hajj, N. DNA Methylation Biomarkers in Aging and Age-Related Diseases. Front. Genet. 2020, 11.
- Tohgi, H.; Utsugisawa, K.; Nagane, Y.; Yoshimura, M.; Ukitsu, M.; Genda, Y. The methylation status of cytosines in a τ gene promoter region alters with age to downregulate transcriptional activity in human cerebral cortex. Neurosci. Lett. 1999, 275, 89–92.
- Tohgi, H.; Utsugisawa, K.; Nagane, Y.; Yoshimura, M.; Genda, Y.; Ukitsu, M. Reduction with age in methylcytosine in the promoter region -224~-101 of the amyloid precursor protein gene in autopsy human cortex. Mol. Brain Res. 1999, 70, 288–292.
- Nagata, K.; Mano, T.; Murayama, S.; Saido, T.C.; Iwata, A. DNA methylation level of the neprilysin promoter in Alzheimer’s disease brains. Neurosci. Lett. 2018, 670, 8–13.
- Hellström-Lindahl, E.; Ravid, R.; Nordberg, A. Age-dependent decline of neprilysin in Alzheimer’s disease and normal brain: Inverse correlation with Aβ levels. Neurobiol. Aging 2008, 29, 210–221.
- Horvath, S.; Zhang, Y.; Langfelder, P.; Kahn, R.S.; Boks, M.P.M.; van Eijk, K.; van den Berg, L.H.; Ophoff, R.A. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012, 13, R97.
- Marioni, R.E.; Harris, S.E.; Shah, S.; McRae, A.F.; von Zglinicki, T.; Martin-Ruiz, C.; Wray, N.R.; Visscher, P.M.; Deary, I.J. The epigenetic clock and telomere length are independently associated with chronological age and mortality. Int. J. Epidemiol. 2016, 45, 424–432.
- Marioni, R.E.; Shah, S.; McRae, A.F.; Ritchie, S.J.; Muniz-Terrera, G.; Harris, S.E.; Gibson, J.; Redmond, P.; Cox, S.R.; Pattie, A.; et al. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int. J. Epidemiol. 2015, 44, 1388–1396.
- Marioni, R.E.; Shah, S.; McRae, A.F.; Chen, B.H.; Colicino, E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Redmond, P.; Cox, S.R.; et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015, 16.
- Levine, M.E.; Lu, A.T.; Bennett, D.A.; Horvath, S. Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging (Albany. NY). 2015, 7, 1198–1211.
- Degerman, S.; Josefsson, M.; Nordin Adolfsson, A.; Wennstedt, S.; Landfors, M.; Haider, Z.; Pudas, S.; Hultdin, M.; Nyberg, L.; Adolfsson, R. Maintained memory in aging is associated with young epigenetic age. Neurobiol. Aging 2017, 55, 167–171.
- Bahado-Singh, R.O.; Vishweswaraiah, S.; Aydas, B.; Yilmaz, A.; Metpally, R.P.; Carey, D.J.; Crist, R.C.; Berrettini, W.H.; Wilson, G.D.; Imam, K.; et al. Artificial intelligence and leukocyte epigenomics: Evaluation and prediction of late-onset Alzheimer’s disease. PLoS ONE 2021, 16.
- Pellegrini, C.; Pirazzini, C.; Sala, C.; Sambati, L.; Yusipov, I.; Kalyakulina, A.; Ravaioli, F.; Kwiatkowska, K.M.; Durso, D.F.; Ivanchenko, M.; et al. A Meta-Analysis of Brain DNA Methylation Across Sex, Age, and Alzheimer’s Disease Points for Accelerated Epigenetic Aging in Neurodegeneration. Front. Aging Neurosci. 2021, 13.
- Wang, Q.; Chen, Y.; Readhead, B.; Chen, K.; Su, Y.; Reiman, E.M.; Dudley, J.T. Longitudinal data in peripheral blood confirm that PM20D1 is a quantitative trait locus (QTL) for Alzheimer’s disease and implicate its dynamic role in disease progression. Clin. Epigenetics 2020, 12.
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic changes in Alzheimer’s disease: Decrements in DNA methylation. Neurobiol. Aging 2010, 31, 2025–2037.
- Chouliaras, L.; Mastroeni, D.; Delvaux, E.; Grover, A.; Kenis, G.; Hof, P.R.; Steinbusch, H.W.M.; Coleman, P.D.; Rutten, B.P.F.; van den Hove, D.L.A. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 2091–2099.
- Mastroeni, D.; McKee, A.; Grover, A.; Rogers, J.; Coleman, P.D. Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer’s disease. PLoS ONE 2009, 4.
- Gasparoni, G.; Bultmann, S.; Lutsik, P.; Kraus, T.F.J.; Sordon, S.; Vlcek, J.; Dietinger, V.; Steinmaurer, M.; Haider, M.; Mulholland, C.B.; et al. DNA methylation analysis on purified neurons and glia dissects age and Alzheimer’s disease-specific changes in the human cortex. Epigenetics Chromatin 2018, 11.
- Foraker, J.; Millard, S.P.; Leong, L.; Thomson, Z.; Chen, S.; Keene, C.D.; Bekris, L.M.; Yu, C.E.; Fischer, A. The APOE Gene is Differentially Methylated in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 48, 745–755.
- Bertram, L.; Lill, C.M.; Tanzi, R.E. The genetics of alzheimer disease: Back to the future. Neuron 2010, 68, 270–281.
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G.; DeStefano, A.L.; Bis, J.C.; Beecham, G.W.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458.
- Yu, C.C.; Jiang, T.; Yang, A.F.; Du, Y.J.; Wu, M.; Kong, L.H. Epigenetic modulation on tau phosphorylation in Alzheimer’s disease. Neural Plast. 2019, 2019.
- Sayas, C.L.; Ávila, J. GSK-3 and tau: A key duet in alzheimer’s disease. Cells 2021, 10, 721.
- Nicolia, V.; Ciraci, V.; Cavallaro, R.A.; Ferrer, I.; Scarpa, S.; Fuso, A. GSK3β 5’-flanking DNA Methylation and Expression in Alzheimer’s Disease Patients. Curr. Alzheimer Res. 2017, 14.
- Li, L.; Zhang, C.; Zi, X.; Tu, Q.; Guo, K. Epigenetic modulation of Cdk5 contributes to memory deficiency induced by amyloid fibrils. Exp. Brain Res. 2015, 233, 165–173.
- Sanchez-Mut, J.V.; Aso, E.; Heyn, H.; Matsuda, T.; Bock, C.; Ferrer, I.; Esteller, M. Promoter hypermethylation of the phosphatase DUSP22 mediates PKA-dependent TAU phosphorylation and CREB activation in Alzheimer’s disease. Hippocampus 2014.
- Do Carmo, S.; Hanzel, C.E.; Jacobs, M.L.; MacHnes, Z.; Iulita, M.F.; Yang, J.; Yu, L.; Ducatenzeiler, A.; Danik, M.; Breuillaud, L.S.; et al. Rescue of Early bace-1 and Global DNA Demethylation by S-Adenosylmethionine Reduces Amyloid Pathology and Improves Cognition in an Alzheimer’s Model. Sci. Rep. 2016, 6.
- Allen, B.; Pezone, A.; Porcellini, A.; Muller, M.T.; Masternak, M.M. Non-homologous end joining induced alterations in DNA methylation: A source of permanent epigenetic change. Oncotarget 2017, 8, 40359–40372.
- Nobile, V.; Pucci, C.; Chiurazzi, P.; Neri, G.; Tabolacci, E. Dna methylation, mechanisms of FMR1 inactivation and therapeutic perspectives for fragile X syndrome. Biomolecules 2021, 11, 1–17.
- Hou, Y.; Song, H.; Croteau, D.L.; Akbari, M.; Bohr, V.A. Genome instability in Alzheimer disease. Mech. Ageing Dev. 2017, 161, 83–94.
- Mano, T.; Nagata, K.; Nonaka, T.; Tarutani, A.; Imamura, T.; Hashimoto, T.; Bannai, T.; Koshi-Mano, K.; Tsuchida, T.; Ohtomo, R.; et al. Neuron-specific methylome analysis reveals epigenetic regulation and tau-related dysfunction of BRCA1 in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E9645–E9654.
- Suberbielle, E.; Djukic, B.; Evans, M.; Kim, D.H.; Taneja, P.; Wang, X.; Finucane, M.; Knox, J.; Ho, K.; Devidze, N.; et al. DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat. Commun. 2015, 6.
- Ambigapathyy, G.; Zhengy, Z.; Keifer, J. Regulation of BDNF chromatin status and promoter accessibility in a neural correlate of associative learning. Epigenetics 2015, 10, 981–993.
- Nakamura, M.; Kaneko, S.; Dickson, D.W.; Kusaka, H. Aberrant accumulation of BRCA1 in Alzheimer disease and other tauopathies. J. Neuropathol. Exp. Neurol. 2020, 79, 22–33.
- Moh, C.; Kubiak, J.Z.; Bajic, V.P.; Zhu, X.; Smith, M.A.; Lee, H.G. Cell cycle deregulation in the neurons of Alzheimer’s disease. Results Probl. Cell Differ. 2011, 53, 565–576.
- Taher, N.; McKenzie, C.; Garrett, R.; Baker, M.; Fox, N.; Isaacs, G.D. Amyloid-β alters the DNA methylation status of cell-fate genes in an Alzheimer’s disease model. J. Alzheimer’s Dis. 2014, 38, 831–844.
- Li, P.; Marshall, L.; Oh, G.; Jakubowski, J.L.; Groot, D.; He, Y.; Wang, T.; Petronis, A.; Labrie, V. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Nat. Commun. 2019, 10.
- Khan, U.A.; Liu, L.; Provenzano, F.A.; Berman, D.E.; Profaci, C.P.; Sloan, R.; Mayeux, R.; Duff, K.E.; Small, S.A. Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease. Nat. Neurosci. 2014, 17, 304–311.
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128.
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014, 17, 1156–1163.
- Watson, C.T.; Roussos, P.; Garg, P.; Ho, D.J.; Azam, N.; Katsel, P.L.; Haroutunian, V.; Sharp, A.J. Genome-wide12 DNA methylation profiling in the superior temporal gyrus reveals epigenetic signatures associated with Alzheimer’s disease. Genome Med. 2016, 8.
- Fuso, A.; Seminara, L.; Cavallaro, R.A.; D’Anselmi, F.; Scarpa, S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol. Cell. Neurosci. 2005, 28, 195–204.
- Scarpa, S.; Cavallaro, R.A.; D’Anselmi, F.; Fuso, A. Gene silencing through methylation: An epigenetic intervention on Alzheimer disease. J. Alzheimer’s Dis. 2006, 9, 407–414.
- Chan, A.; Shea, T.B. Effects of dietary supplementation with N-acetyl cysteine, acetyl-L-carnitine and S-adenosyl methionine on cognitive performance and aggression in normal mice and mice expressing human ApoE4. NeuroMolecular Med. 2008, 10, 46.
- Fuso, A. The “golden age” of DNA methylation in neurodegenerative diseases. Proc. Clin. Chem. Lab. Med. 2013, 51, 523–534.
- Fuso, A.; Scarpa, S. One-carbon metabolism and Alzheimer’s disease: Is it all a methylation matter? Neurobiol. Aging 2011, 32, 1192–1195.
- Momparler, R.L.; Momparler, L.F.; Samson, J. Comparison of the antileukemic activity of 5-aza-2′-deoxycytidine, 1-β-d-arabinofuranosylcytosine and 5-azacytidine against L1210 leukemia. Leuk. Res. 1984, 8, 1043–1049.
- Issa, J.P.J.; Garcia-Manero, G.; Giles, F.J.; Mannari, R.; Thomas, D.; Faderl, S.; Bayar, E.; Lyons, J.; Rosenfeld, C.S.; Cortes, J.; et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood 2004, 103, 1635–1640.
- Lübbert, M.; Daskalakis, M.; Sander, P.N.; Kündgen, A. Azanucleoside DNA Methyltransferase inhibitor drugs: Update on clinical applications in Myelodysplastic syndromes and acute myeloid leukemia. In Proceedings of the Epigenetics and Human Health; Springer Nature: Cham, Switzerland, 2016; pp. 197–221.
- Sandi, C.; Sandi, M.; Virmouni, S.A.; Al-Mahdawi, S.; Pook, M.A. Epigenetic-based therapies for Friedreich ataxia. Front. Genet. 2014, 5.
- Plummer, R.; Vidal, L.; Griffin, M.; Lesley, M.; De Bono, J.; Coulthard, S.; Sludden, J.; Siu, L.L.; Chen, E.X.; Oza, A.M.; et al. Phase I study of MG98, an oligonucleotide antisense inhibitor of human DNA methyltransferase 1, given as a 7-day infusion in patients with advanced solid tumors. Clin. Cancer Res. 2009, 15, 3177–3183.
- Winquist, E.; Knox, J.; Ayoub, J.P.; Wood, L.; Wainman, N.; Reid, G.K.; Pearce, L.; Shah, A.; Eisenhauer, E. Phase II trial of DNA methyltransferase 1 inhibition with the antisense oligonucleotide MG98 in patients with metastatic renal carcinoma: A National Cancer Institute of Canada Clinical Trials Group investigational new drug study. Investig. New Drugs 2006, 24, 159–167.
- Stewart, D.J.; Donehower, R.C.; Eisenhauer, E.A.; Wainman, N.; Shah, A.K.; Bonfils, C.; MacLeod, A.R.; Besterman, J.M.; Reid, G.K. A phase I pharmacokinetic and pharmacodynamic study of the DNA methyltransferase 1 inhibitor MG98 administered twice weekly. Ann. Oncol. 2003, 14, 766–774.
- Urbano, A.; Smith, J.; Weeks, R.J.; Chatterjee, A. Gene-specific targeting of DNA methylation in the mammalian genome. Cancers 2019, 11, 1515.
- Xie, B.; Xu, Y.; Liu, Z.; Liu, W.; Jiang, L.; Zhang, R.; Cui, D.; Zhang, Q.; Xu, S. Elevation of peripheral BDNF promoter methylation predicts conversion from amnestic mild cognitive impairment to Alzheimer’s disease: A 5-year longitudinal study. J. Alzheimer’s Dis. 2017, 56, 391–401.
- Kobayashi, N.; Shinagawa, S.; Nagata, T.; Shimada, K.; Shibata, N.; Ohnuma, T.; Kasanuki, K.; Arai, H.; Yamada, H.; Nakayama, K.; et al. Usefulness of DNA methylation levels in COASY and SPINT1 gene promoter regions as biomarkers in diagnosis of Alzheimer’s disease and Amnestic mild cognitive impairment. PLoS ONE 2016, 11.
- Mercorio, R.; Pergoli, L.; Galimberti, D.; Favero, C.; Carugno, M.; Dalla Valle, E.; Barretta, F.; Cortini, F.; Scarpini, E.; Valentina, V.B.; et al. PICALM gene methylation in blood of Alzheimer’s disease patients is associated with cognitive decline. J. Alzheimer’s Dis. 2018, 65, 283–292.
- Di Francesco, A.; Arosio, B.; Falconi, A.; Micioni Di Bonaventura, M.V.; Karimi, M.; Mari, D.; Casati, M.; Maccarrone, M.; D’Addario, C. Global changes in DNA methylation in Alzheimer’s disease peripheral blood mononuclear cells. Brain. Behav. Immun. 2015, 45, 139–144.