With a rapidly growing elderly human population, the incidence of age-related lung diseases such as chronic obstructive pulmonary disease (COPD) continues to rise. COPD is a chronic irreversible disease of the lungs characterized by airflow limitation due to destruction of the lung parenchyma (emphysema) and/or remodelling of the small airways, and is currently the third leading cause of death in the Westernized world. The greatest risk factor for COPD is smoking, but not all smokers develop COPD and the reasons for disease susceptibility in these individuals remains poorly understood. In COPD, the alveolar architecture has been destroyed resulting in emphysemaand subsequent dyspnea (shortness of breath).
- ageing
- COPD
1. Introduction
Human life expectancy has nearly doubled globally during the past century, and the global human population over the age of 65 is expected to represent ~20% of the world’s population by 2050 [1]. Ageing is characterized by gradual and irreversible functional deterioration of all vital organs after the reproductive phase of life is complete [2], and is a major risk factor for death from all adult chronic diseases. Therefore, the rapid increase in the ageing population together with declining fertility rates will create an ever-increasing societal burden and health care challenge over the next decades, and demands for increased understanding of the molecular mechanisms underlying ageing and age-related disease, enabling advanced health care for our elderly, will become increasingly urgent.
On a cellular and molecular level, Lopez-Otín and colleagues have defined nine hallmarks of ageing, which include genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication [3]. Dysregulation of the extracellular matrix due to aging is an additional crucial modifier of cell-autonomous changes and functions [4]. The origin of these various hallmarks is undoubtedly complex, and is likely to involve a combination of underlying processes that may be cell- and organ-specific, and the individual contribution of each hallmark to individual ageing-related non-communicable chronic diseases may vary.
One well-recognized aspect of ageing is the enhanced production of reactive oxygen species (ROS), which are generated during cellular metabolism of molecular O 2 and lead to accumulation of biomolecular oxidative damage [5][6][7]. This, combined with the lifelong exposure to ionizing radiation or environmental oxidizing pollutants, has led Denham Harman to propose the free radical theory (FRT) of ageing [8]. This theory was later refined to the mitochondrial free radical theory of ageing, based on the fact that mitochondria are the primary source of ROS, and in line with mitochondrial dysfunction as one of the main hallmarks of ageing [3][9]. Evolutionary evidence does not always support the FRT of ageing, however, and the recognition of physiological functions of ROS in e.g., host defense and other aspects of cell biology through redox-based signalling has further complicated the FRT of ageing. Indeed, the recently discovered family of NADPH oxidase (NOX) enzymes are critical in these physiological roles, and the association(s) between NOX function and redox-based signalling and ageing are only beginning to be appreciated (e.g., [10][11]).
The lung is the organ with the largest surface area that faces the external environment, estimated to be as large as half a tennis court [12], and is therefore exceptionally vulnerable to the life-long exposure to environmental pathogens and common (oxidizing) airborne pollutants. Indeed, ageing is associated with a progressive decline in lung function and with increased susceptibility to the development of chronic age-related pulmonary diseases, such as chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF), all rapidly increasing in incidence with advancing age [13][14]. Likewise, oxidative stress (due elevated levels of ROS and/or impaired antioxidant defenses) is often viewed as a common feature of, and contributor to these diseases [15], and has encouraged the proposed use of antioxidant-based strategies in potential treatment of these diseases.
2. The Ageing Lung
Throughout human lifespan, various age-associated structural and functional changes occur within the respiratory system, termed the lung function trajectories. Lung growth occurs from birth until adulthood and is characterized by increases in lung volume, an increase in the number of alveoli, and increased capillary networks [16]. During the plateau phase (adolescence ~25 years of age to 30–40 years) these numbers remain stable [17], after which [18], lung function starts to gradually decline with increasing age, which may be variable in every individual based on genetics and different exposure histories to e.g., cigarette smoke or other environmental challenges. It is important to recognize that individual lung growth may vary as well, and that abnormal lung growth early in life may also affect later phases of lung function trajectories [19][20].
The decline phase has various consequences for functional capacity in absence of underlying pathology and affects every individual, with early limitations only observed during exercise and later on during broader settings. Accordingly, with advancing age, the respiratory tract undergoes both structural and physiological changes, such as loss of lung regenerative capacity and pulmonary remodelling, which are associated with a progressive decrease in lung function [21]. Characteristic of the ageing lung is a decrease in lung elasticity and concomitant increase in alveolar size. This loss in lung elasticity and airway enlargement results in increased functional residual capacity (FRC) and end-expiratory lung volume (EELV). Additionally, the ratio between the forced expiratory volume in one second and forced vital capacity (FEV1/FVC), often used to diagnose chronic obstructive lung diseases and defined as the amount of air that can be forcibly exhaled following one’s maximal inhalation, decreases with age due to loss of lung elasticity and airspace enlargement, and also because of loss of respiratory muscle mass [17]. Also termed the ‘senile lung’, these age-related structural lung changes are mainly attributed to an increase in the size of the alveolar space and are not considered pathological because they occur in the absence of significant inflammation or alveolar wall destruction.
In addition to negatively impacting lung structure and physiology, ageing is known to lead to a gradual dysregulation of the immune system, which is characterized by an impaired ability of various immune cells to respond to pathogens, and by age-related low-grade inflammation due to immunosenescence (known as inflammageing) [22][23][24]. Replicative and/or stress-induced cellular senescence of immune cells results in compromised and inappropriate cellular function and cell responses of e.g., innate and humoral immunity [25]. This is largely responsible for the increased susceptibility of elderly subjects to infection with influenza virus or with SARS-CoV-2 [26][27][28]. Replicative senescence of resident cells induces the senescence-associated secretory phenotype (SASP), which is characterized by resident senescent cells secreting pro-inflammatory factors that can alter the cellular microenvironment and shift neighbouring healthy proliferating cells into a more senescent- and pro-inflammatory state. In addition to damage-associated molecular patterns (DAMPs), the SASP contributes to inflammageing/sterile inflammation observed during lung ageing [29], which is characterized by pro-inflammatory cytokine release and chronic low-grade inflammation in the absence of an immunological threat [30]. While associated with ageing, the SASP has likely evolved as a mechanism to maintain homeostasis through senescent cell clearance, progenitor cell repopulation, and wound healing and tissue repair [25][31][32], and has also been shown to counter early-life tumorigenesis [33].
Because the respiratory system represents a critical interface with the external environment and is susceptible to injury from inhaled environmental pathogens and pollutants, it is equipped with various defense mechanisms (antioxidant defenses, antimicrobial defenses, mucus and mucociliary clearance mechanisms, and local sentinel immune cells). Age-related decline in these mechanisms likely contributes to biochemical and physiological changes in the lung, and may contribute to the development of age-related chronic lung disease(s) [34][35][36]. Indeed, while the senile lung is characterized by airspace enlargement in the absence of overt inflammation and tissue remodelling, such compromised and inappropriate responses to exogenous hazards in the ageing lung likely contribute to chronic inflammation and alveolar wall destruction that contribute to the development of e.g., emphysema [37][38], and also render the aged lung more susceptible to acute injury or infections that contribute to exacerbations, and may in turn further aggravate lung ageing. Hence, the molecular mechanisms underlying chronic lung diseases such as COPD are also dictated by alterations that occur as a result of normal aging. Recent efforts using single-cell transcriptomics and proteomics to develop an atlas of normal aging, such as the Tabula Muris Senis database [39], have dramatically increased our insights into (lung) cell-type specific effects of ageing [40], and present highly useful resources to assess the contributions of ageing to chronic age-related diseases, including those of the lung.
3. COPD a Disease of Accelerated Ageing?
COPD is a chronic irreversible disease of the lungs characterized by airflow limitation due to destruction of the lung parenchyma (emphysema) and/or remodelling of the small airways, and is currently the third leading cause of death in the Westernized world [41]. The greatest risk factor for COPD is smoking, but not all smokers develop COPD and the reasons for disease susceptibility in these individuals remains poorly understood [42]. In COPD, the alveolar architecture has been destroyed resulting in emphysema [43][44] and subsequent dyspnea (shortness of breath). Small airway disease and emphysema development are mechanistically related, since small airway inflammation may propagate to the alveolar septa, in turn destroying bronchiolar-alveolar attachments, and eventually proceed into lung parenchymal destruction [45][46][47]. Moreover, a loss of small airways before the onset of parenchymal destruction may explain the increased peripheral airway resistance described in COPD [44]. Another histopathological feature often observed in COPD patients is seen in the vasculature with increased thickness of the arterioles, resulting in pulmonary hypertension as an additional complication of COPD [48].
Various genetic factors have been established as risk factors for COPD, such as genetic defects in the SERPIN1 gene resulting in alpha1-antitrypsin deficiency [49][50]. The most widely recognized cause of COPD pathogenesis is however exposure to repeated environmental insults such as tobacco smoke, which is associated with repetitive injury and persistent inflammation and imbalanced protease/anti-protease activities within the lung [51][52], thus leading to progressive lung tissue damage, abnormal tissue remodelling, and tissue fibrosis [53]. As a result, COPD is characterized primarily by thickening of (large and small) airways due to subepithelial fibrosis and mucus plugging, and a related obstruction of the small airway lumen, and with alveolar emphysema due to alveolar wall destruction and loss of alveolar surface area.
At the cellular and molecular level, COPD is characterized by various alterations of cell biology such as telomere shortening [54][55] and senescence/SASP in many cell types [56], including endothelial [57] and (alveolar and bronchial) epithelial cells [58], as well as fibroblasts [59]. Furthermore, COPD is characterized by altered/impaired innate immune function that may contribute to infection and exacerbations in this disease. These various alterations and decline in function are greatly impacted by cigarette smoking [54][58]. For example, cigarette smoking may contribute to basal cell hyperplasia as one of the initial events of altered epithelial cell biology in COPD [60]. Such alterations in the basal cell population also contribute to airway epithelial remodelling phenotypes including mucous cell hyperplasia, epithelial-mesenchymal transition (EMT), altered cell differentiation, and impaired epithelial barrier integrity [61][62].
More recent transcriptional profiling studies of airway basal cells from COPD patients and non-COPD controls revealed a marked heterogeneity indicating a continuum of basal cell status that may represent gradually evolving trajectories of basal cell phenotypes as COPD develops [63]. Transcriptional analyses also indicated that smoking can induce a distal-to-proximal repatterning of small airway epithelial cells, which was attributed to increased activation of the epidermal growth factor (EGF)/epidermal growth factor receptor (EGFR) pathways [64]. Transcriptional analysis also suggested a reprogramming of alveolar macrophages in COPD, with relatively less M1 polarization and a shift towards partial M2 polarization. These alterations appear to correlate with COPD severity [65], and to be driven by oxidative stress induced by smoking, as they lead to impaired innate macrophage activation in response to e.g., infection [66][67]. Mechanistic studies suggested a potential for acrolein, a major electrophile of CS, in such macrophage alterations, due to the reactivity of acrolein towards thiols within critical proteins involved in macrophage activation/polarization such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and c-Jun N-terminal kinase 2 (JNK2) [68]. COPD may develop through variable lung function trajectories. Indeed, while some COPD patients may display accelerated age-related lung function decline following normal lung growth, others show evidence of abnormal lung growth with normal age-related lung function decline [19][20]. Furthermore, the ageing lung is characterized by ‘senile emphysema’, which is characterized by a loss of elasticity, enlargement of alveoli as well as low-grade inflammation. However, it is not a result of destruction of the alveolar walls, which does underlie emphysema in COPD. Cellular senescence is observed during lung ageing and may suggest a predisposition to COPD development. As such, examining and understanding the underlying molecular mechanisms involved in normal lung ageing (e.g., based on available insights from public databases such as Tabula Muris Senis) may help to understand how tobacco smoke and other oxidative stressors may accelerate lung ageing and result in COPD development. Indeed, many of the known hallmarks of ageing are also thought to contribute to COPD pathogenesis, such as epigenetic alterations (e.g., due to dysregulation of histone deacetylases), loss of proteostasis (regulation of protein biogenesis, folding, trafficking and degradation), mitochondrial dysfunction, and cellular senescence [3][4]. Altered intercellular communication (e.g., adaptive immune responses), and abnormal extracellular matrix (ECM) turnover and deposition further contribute to COPD pathogenesis [4].
4. NOX Enzymes in COPD Pathology
Similar to the relative lack of available studies addressing associations of NOX enzymes with ageing, a rather limited number of previous studies have attempted to address the specific role(s) of NOX enzymes in COPD pathology. Not surprisingly, increased numbers of NOX2-positive inflammatory cells have been observed in lung tissues from COPD patients, and a contributing role of macrophage NOX2 in elastase-induced emphysema has been reported using NOX2-deficient mice [69]. However, while NOX2 contributes to macrophage-mediated oxidative stress and inflammation due to cigarette smoke exposure [70], genetic deletion of NOX2 in mice was actually found to aggravate CS-induced emphysema, which was associated with increased inflammation that was perhaps worsened due to NOX2 deficiency [71][72]. These discrepant findings may be related to the different animal models used, variable roles of NOX2 in limiting chronic inflammation through e.g., Nrf2 [71] or promoting injury during acute inflammation (e.g., in the case of the elastase model), and diverse functions of NOX2 in different cell types, which would be best dissected using cell- or tissue-specific NOX2 knockout strategies. Some studies have reported elevated levels of NOX4 in airway smooth muscle of COPD patients [73], which were found to correlate with disease severity [74] and to be associated with pulmonary hypertension [75]. Furthermore, RTP801/REDD1, which negatively regulates mammalian target of rapamycin (mTOR), is upregulated in response to cigarette smoke, and enhances inflammation and alveolar destruction by increasing NOX4 activity [76]. In a genetic mouse model of emphysema (due to TLR4 deficiency), elastolytic activity was found to be increased due to induction of NOX3 in the pulmonary endothelium and resultant oxidant generation [77], but the relevance for human COPD is unclear.
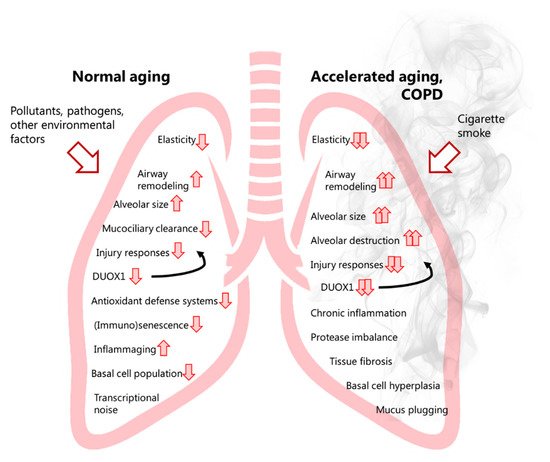
Analysis of tracheal and bronchial epithelium collected by airway brushing or laser capture micro-dissection, revealed that DUOX1 was significantly suppressed in the airways of healthy smokers and patients with COPD, when compared to age-matched control subjects, implying that chronic smoking leads to decreased airway epithelial expression of DUOX1 as a potential contributing factor in COPD development [78][79]. Our group expanded on these findings, by showing a gradual downregulation of DUOX1 protein in the small airways of GOLD II-IV COPD patients, which was found to be strongly correlated with various measures of lung function decline, and several markers of small airway remodeling and destruction [80]. On one hand, these results may imply that variable DUOX1 downregulation as a result of normal aging (see above) or smoking history may predispose for COPD development and progression. Alternatively, it is also possible that DUOX1 downregulation may be a consequence of COPD, for example secondary to production of inflammatory mediators or growth factors such as TGF-β. Indeed, we observed that chronic exposure of bronchial epithelial cells to TGF-β also downregulates DUOX1 [80]. Downregulation of DUOX1 may be related to smoking history, although some studies suggest that exposure to CS extract can actually enhance DUOX1 expression [81]. In contrast, chronic exposure of mice to the CS-component acrolein was found to result in DUOX1 downregulation, a response that could be mimicked by chronic in vitro exposure of epithelial cells to acrolein [80]. Nevertheless, the relationship between airway or alveolar DUOX1 and smoking status/history is complex, and observed correlations of airway DUOX1 with lung function parameters in our recent studies were largely independent of smoking status [80], suggesting the contribution of other factors to DUOX1 downregulation in COPD. To address a potential causal effect of DUOX1 down-regulation in COPD development or progression, we assessed the impact of DUOX1 deletion in a mouse model of elastase-induced emphysema or in a mouse model of small airway remodeling due to chronic acrolein exposure. In both cases, we observed worse disease phenotypes in DUOX1-deficient mice suggesting that DUOX1 down-regulation in COPD may actively contribute to disease progression, likely related to altered epithelial biology and homeostasis, as well as neutrophilic inflammation and degranulation [80]. Moreover, in light of recent work indicating a role for DUOX1 in antiviral innate immunity [82], decreased DUOX1 in the lung of COPD patients may also promote susceptibility to viral infection and may thereby enhance exacerbations [83]. Overall, the apparent roles of NOX enzymes in COPD pathology are variable, with some NOX enzymes (e.g., NOX2 and NOX4) contributing to aspects of COPD pathogenesis, whereas others (most notably DUOX1, Figure 1 ) may actually help to prevent COPD progression.
This entry is adapted from the peer-reviewed paper 10.3390/antiox10111799
References
- Dzau, V.J.; Inouye, S.K.; Rowe, J.W.; Finkelman, E.; Yamada, T. Enabling healthful aging for all—The national academy of medicine grand challenge in healthy longevity. N. Engl. J. Med. 2019, 381, 1699–1701.
- Mangel, M. Complex adaptive systems, aging and longevity. J. Theor. Biol. 2001, 213, 559–571.
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217.
- Meiners, S.; Eickelberg, O.; Konigshoff, M. Hallmarks of the ageing lung. Eur. Respir. J. 2015, 45, 807–827.
- Freeman, B.A.; Crapo, J.D. Biology of disease: Free radicals and tissue injury. Lab. Investig. 1982, 47, 412–426.
- Halliwell, B. The wanderings of a free radical. Free Radic. Biol. Med. 2009, 46, 531–542.
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95.
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300.
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495.
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; et al. A quantitative tissue-specific landscape of protein redox regulation during aging. Cell 2020, 180, 968–983.
- Krause, K.H. Aging: A revisited theory based on free radicals generated by NOX family NADPH oxidases. Exp. Gerontol. 2007, 42, 256–262.
- Frohlich, E.; Mercuri, A.; Wu, S.; Salar-Behzadi, S. Measurements of deposition, lung surface area and lung fluid for simulation of inhaled compounds. Front. Pharmacol. 2016, 7, 181.
- Budinger, G.R.S.; Kohanski, R.A.; Gan, W.; Kobor, M.S.; Amaral, L.A.; Armanios, M.; Kelsey, K.T.; Pardo, A.; Tuder, R.; Macian, F.; et al. The intersection of aging biology and the pathobiology of lung diseases: A joint nhlbi/nia workshop. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2017, 72, 1492–1500.
- Ito, K.; Barnes, P.J. COPD as a disease of accelerated lung aging. Chest 2009, 135, 173–180.
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772.
- Schittny, J.C. Development of the lung. Cell Tissue Res. 2017, 367, 427–444.
- Bowdish, D.M.E. The aging lung: Is lung health good health for older adults? Chest 2019, 155, 391–400.
- Fletcher, C.; Peto, R. The natural history of chronic airflow obstruction. BMJ 1977, 1, 1645–1648.
- Agusti, A.; Faner, R. Lung function trajectories in health and disease. Lancet Respir. Med. 2019, 7, 358–364.
- Agusti, A.; Hogg, J.C. Update on the pathogenesis of chronic obstructive pulmonary disease. N. Engl. J. Med. 2019, 381, 1248–1256.
- Brandenberger, C.; Muhlfeld, C. Mechanisms of lung aging. Cell Tissue Res. 2017, 367, 469–480.
- Cho, S.J.; Stout-Delgado, H.W. Aging and lung disease. Annu. Rev. Physiol. 2020, 82, 433–459.
- Schneider, J.L.; Rowe, J.H.; Garcia-de-Alba, C.; Kim, C.F.; Sharpe, A.H.; Haigis, M.C. The aging lung: Physiology, disease, and immunity. Cell 2021, 184, 1990–2019.
- Shaw, A.C.; Goldstein, D.R.; Montgomery, R.R. Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 2013, 13, 875–887.
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496.
- Farshbafnadi, M.; Kamali Zonouzi, S.; Sabahi, M.; Dolatshahi, M.; Aarabi, M.H. Aging & COVID-19 susceptibility, disease severity, and clinical outcomes: The role of entangled risk factors. Exp. Gerontol. 2021, 154, 111507.
- Bajaj, V.; Gadi, N.; Spihlman, A.P.; Wu, S.C.; Choi, C.H.; Moulton, V.R. Aging, immunity, and COVID-19: How age influences the host immune response to coronavirus infections? Front. Physiol. 2020, 11, 571416.
- Chen, Y.; Klein, S.L.; Garibaldi, B.T.; Li, H.; Wu, C.; Osevala, N.M.; Li, T.; Margolick, J.B.; Pawelec, G.; Leng, S.X. Aging in COVID-19: Vulnerability, immunity and intervention. Ageing Res. Rev. 2021, 65, 101205.
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. S1), S4–S9.
- Panda, A.; Arjona, A.; Sapey, E.; Bai, F.; Fikrig, E.; Montgomery, R.R.; Lord, J.M.; Shaw, A.C. Human innate immunosenescence: Causes and consequences for immunity in old age. Trends Immunol. 2009, 30, 325–333.
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733.
- Sagiv, A.; Krizhanovsky, V. Immunosurveillance of senescent cells: The bright side of the senescence program. Biogerontology 2013, 14, 617–628.
- Ghosh, K.; Capell, B.C. The senescence-associated secretory phenotype: Critical effector in skin cancer and aging. J. Investig. Dermatol. 2016, 136, 2133–2139.
- Wang, L.; Green, F.H.; Smiley-Jewell, S.M.; Pinkerton, K.E. Susceptibility of the aging lung to environmental injury. Semin. Respir. Crit. Care Med. 2010, 31, 539–553.
- Bustos, M.L.; Huleihel, L.; Kapetanaki, M.G.; Lino-Cardenas, C.L.; Mroz, L.; Ellis, B.M.; McVerry, B.J.; Richards, T.J.; Kaminski, N.; Cerdenes, N.; et al. Aging mesenchymal stem cells fail to protect because of impaired migration and antiinflammatory response. Am. J. Respir. Crit. Care. Med. 2014, 189, 787–798.
- Katial, R.; Zheng, W. Allergy and immunology of the aging lung. Clin. Chest Med. 2007, 28, 663–672.
- Verbeken, E.K.; Cauberghs, M.; Mertens, I.; Clement, J.; Lauweryns, J.M.; Van de Woestijne, K.P. The senile lung. Comparison with normal and emphysematous lungs. 1. Structural aspects. Chest 1992, 101, 793–799.
- Verbeken, E.K.; Cauberghs, M.; Mertens, I.; Clement, J.; Lauweryns, J.M.; Van de Woestijne, K.P. The senile lung. Comparison with normal and emphysematous lungs. 2. Functional aspects. Chest 1992, 101, 800–809.
- Tabula Muris, C. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 2020, 583, 590–595.
- Angelidis, I.; Simon, L.M.; Fernandez, I.E.; Strunz, M.; Mayr, C.H.; Greiffo, F.R.; Tsitsiridis, G.; Ansari, M.; Graf, E.; Strom, T.M.; et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat. Commun. 2019, 10, 963.
- Quaderi, S.A.; Hurst, J.R. The unmet global burden of COPD. Glob. Health Epidemiol. Genom. 2018, 3, e4.
- Barnes, P.J.; Burney, P.G.; Silverman, E.K.; Celli, B.R.; Vestbo, J.; Wedzicha, J.A.; Wouters, E.F. Chronic obstructive pulmonary disease. Nat. Rev. Dis. Primers 2015, 1, 15076.
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med. 2004, 350, 2645–2653.
- McDonough, J.E.; Yuan, R.; Suzuki, M.; Seyednejad, N.; Elliott, W.M.; Sanchez, P.G.; Wright, A.C.; Gefter, W.B.; Litzky, L.; Coxson, H.O.; et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N. Engl. J. Med. 2011, 365, 1567–1575.
- Janssen, R.; Wouters, E.F.M. Loss of alveolar attachments as a pathomechanistic link between small airway disease and emphysema. Am. J. Respir. Crit. Care. Med. 2020, 201, 878–879.
- Kirby, M.; Tanabe, N.; Tan, W.C.; Zhou, G.; Obeidat, M.; Hague, C.J.; Leipsic, J.; Bourbeau, J.; Sin, D.D.; Hogg, J.C.; et al. Total airway count on computed tomography and the risk of chronic obstructive pulmonary disease progression. Findings from a population-based study. Am. J. Respir. Crit. Care. Med. 2018, 197, 56–65.
- Mitzner, W. Emphysema—A disease of small airways or lung parenchyma? N. Engl. J. Med. 2011, 365, 1637–1639.
- Berg, K.; Wright, J.L. The pathology of chronic obstructive pulmonary disease: Progress in the 20th and 21st centuries. Arch. Pathol. Lab. Med. 2016, 140, 1423–1428.
- Hall, R.; Hall, I.P.; Sayers, I. Genetic risk factors for the development of pulmonary disease identified by genome-wide association. Respirology 2019, 24, 204–214.
- Silverman, E.K. Genetics of COPD. Annu. Rev. Physiol. 2020, 82, 413–431.
- Rahman, I.; Adcock, I.M. Oxidative stress and redox regulation of lung inflammation in COPD. Eur. Respir. J. 2006, 28, 219–242.
- Hogg, J.C.; Timens, W. The pathology of chronic obstructive pulmonary disease. Annu. Rev. Pathol. 2009, 4, 435–459.
- Brandsma, C.A.; De Vries, M.; Costa, R.; Woldhuis, R.R.; Konigshoff, M.; Timens, W. Lung ageing and COPD: Is there a role for ageing in abnormal tissue repair? Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2017, 26, 170073.
- Morla, M.; Busquets, X.; Pons, J.; Sauleda, J.; MacNee, W.; Agusti, A.G. Telomere shortening in smokers with and without COPD. Eur. Respir. J. 2006, 27, 525–528.
- Rutten, E.P.; Gopal, P.; Wouters, E.F.; Franssen, F.M.; Hageman, G.J.; Vanfleteren, L.E.; Spruit, M.A.; Reynaert, N.L. Various mechanistic pathways representing the aging process are altered in COPD. Chest 2016, 149, 53–61.
- Parikh, P.; Wicher, S.; Khandalavala, K.; Pabelick, C.M.; Britt, R.D.; Prakash, Y.S., Jr. Cellular senescence in the lung across the age spectrum. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L826–L842.
- Paschalaki, K.E.; Starke, R.D.; Hu, Y.; Mercado, N.; Margariti, A.; Gorgoulis, V.G.; Randi, A.M.; Barnes, P.J. Dysfunction of endothelial progenitor cells from smokers and chronic obstructive pulmonary disease patients due to increased DNA damage and senescence. Stem Cells 2013, 31, 2813–2826.
- Tsuji, T.; Aoshiba, K.; Nagai, A. Cigarette smoke induces senescence in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 31, 643–649.
- Woldhuis, R.R.; Heijink, I.H.; Van den Berge, M.; Timens, W.; Oliver, B.G.G.; De Vries, M.; Brandsma, C.A. COPD-derived fibroblasts secrete higher levels of senescence-associated secretory phenotype proteins. Thorax 2021, 76, 508–511.
- Shaykhiev, R.; Crystal, R.G. Early events in the pathogenesis of chronic obstructive pulmonary disease. Smoking-induced reprogramming of airway epithelial basal progenitor cells. Ann. Am. Thorac. Soc. 2014, 11 (Suppl. S5), S252–S258.
- Hiemstra, P.S.; McCray, P.B.; Bals, R., Jr. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur. Respir. J. 2015, 45, 1150–1162.
- Puchelle, E.; Zahm, J.M.; Tournier, J.M.; Coraux, C. Airway epithelial repair, regeneration, and remodeling after injury in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2006, 3, 726–733.
- Wijk, S.C.; Prabhala, P.; Michalikova, B.; Sommarin, M.; Doyle, A.; Lang, S.; Kanzenbach, K.; Tufvesson, E.; Lindstedt, S.; Leigh, N.D.; et al. Human primary airway basal cells display a continuum of molecular phases from health to disease in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2021, 65, 103–113.
- Yang, J.; Zuo, W.L.; Fukui, T.; Chao, I.; Gomi, K.; Lee, B.; Staudt, M.R.; Kaner, R.J.; Strulovici-Barel, Y.; Salit, J.; et al. Smoking-dependent distal-to-proximal repatterning of the adult human small airway epithelium. Am. J. Respir. Crit. Care Med. 2017, 196, 340–352.
- Bazzan, E.; Turato, G.; Tine, M.; Radu, C.M.; Balestro, E.; Rigobello, C.; Biondini, D.; Schiavon, M.; Lunardi, F.; Baraldo, S.; et al. Dual polarization of human alveolar macrophages progressively increases with smoking and COPD severity. Respir. Res. 2017, 18, 40.
- Doyle, I.; Ratcliffe, M.; Walding, A.; Vanden Bon, E.; Dymond, M.; Tomlinson, W.; Tilley, D.; Shelton, P.; Dougall, I. Differential gene expression analysis in human monocyte-derived macrophages: Impact of cigarette smoke on host defence. Mol. Immunol. 2010, 47, 1058–1065.
- Shaykhiev, R.; Krause, A.; Salit, J.; Strulovici-Barel, Y.; Harvey, B.G.; O’Connor, T.P.; Crystal, R.G. Smoking-dependent reprogramming of alveolar macrophage polarization: Implication for pathogenesis of chronic obstructive pulmonary disease. J. Immunol. 2009, 183, 2867–2883.
- Hristova, M.; Spiess, P.C.; Kasahara, D.I.; Randall, M.J.; Deng, B.; Van der Vliet, A. The tobacco smoke component, acrolein, suppresses innate macrophage responses by direct alkylation of c-Jun N-terminal kinase. Am. J. Respir. Cell Mol. Biol. 2012, 46, 23–33.
- Trocme, C.; Deffert, C.; Cachat, J.; Donati, Y.; Tissot, C.; Papacatzis, S.; Braunersreuther, V.; Pache, J.C.; Krause, K.H.; Holmdahl, R.; et al. Macrophage-specific NOX2 contributes to the development of lung emphysema through modulation of SIRT1/MMP-9 pathways. J. Pathol. 2015, 235, 65–78.
- Tollefson, A.K.; Oberley-Deegan, R.E.; Butterfield, K.T.; Nicks, M.E.; Weaver, M.R.; Remigio, L.K.; Decsesznak, J.; Chu, H.W.; Bratton, D.L.; Riches, D.W.; et al. Endogenous enzymes (NOX and ECSOD) regulate smoke-induced oxidative stress. Free Radic. Biol. Med. 2010, 49, 1937–1946.
- Singel, K.L.; Segal, B.H. NOX2-dependent regulation of inflammation. Clin. Sci. 2016, 130, 479–490.
- Yao, H.; Edirisinghe, I.; Yang, S.R.; Rajendrasozhan, S.; Kode, A.; Caito, S.; Adenuga, D.; Rahman, I. Genetic ablation of NADPH oxidase enhances susceptibility to cigarette smoke-induced lung inflammation and emphysema in mice. Am. J. Pathol. 2008, 172, 1222–1237.
- Hollins, F.; Sutcliffe, A.; Gomez, E.; Berair, R.; Russell, R.; Szyndralewiez, C.; Saunders, R.; Brightling, C. Airway smooth muscle NOX4 is upregulated and modulates ROS generation in COPD. Respir. Res. 2016, 17, 84.
- Liu, X.; Hao, B.; Ma, A.; He, J.; Liu, X.; Chen, J. The expression of NOX4 in smooth muscles of small airway correlates with the disease severity of COPD. Biomed. Res. Int. 2016, 2016, 2891810.
- Guo, X.; Fan, Y.; Cui, J.; Hao, B.; Zhu, L.; Sun, X.; He, J.; Yang, J.; Dong, J.; Wang, Y.; et al. NOX4 expression and distal arteriolar remodeling correlate with pulmonary hypertension in COPD. BMC Pulm. Med. 2018, 18, 111.
- Hernandez-Saavedra, D.; Sanders, L.; Perez, M.J.; Kosmider, B.; Smith, L.P.; Mitchell, J.D.; Yoshida, T.; Tuder, R.M. RTP801 amplifies nicotinamide adenine dinucleotide phosphate oxidase-4-dependent oxidative stress induced by cigarette smoke. Am. J. Respir. Cell Mol. Biol. 2017, 56, 62–73.
- Zhang, X.; Shan, P.; Jiang, G.; Cohn, L.; Lee, P.J. Toll-like receptor 4 deficiency causes pulmonary emphysema. J. Clin. Investig. 2006, 116, 3050–3059.
- Nagai, K.; Betsuyaku, T.; Suzuki, M.; Nasuhara, Y.; Kaga, K.; Kondo, S.; Nishimura, M. Dual oxidase 1 and 2 expression in airway epithelium of smokers and patients with mild/moderate chronic obstructive pulmonary disease. Antioxid. Redox Signal. 2008, 10, 705–714.
- Pierrou, S.; Broberg, P.; O’Donnell, R.A.; Pawlowski, K.; Virtala, R.; Lindqvist, E.; Richter, A.; Wilson, S.J.; Angco, G.; Moller, S.; et al. Expression of genes involved in oxidative stress responses in airway epithelial cells of smokers with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2007, 175, 577–586.
- Schiffers, C.; Van de Wetering, C.; Bauer, R.A.; Habibovic, A.; Hristova, M.; Dustin, C.M.; Lambrichts, S.; Vacek, P.M.; Wouters, E.F.; Reynaert, N.L.; et al. Downregulation of epithelial DUOX1 in chronic obstructive pulmonary disease. JCI Insight 2021, 6.
- Tian, Z.; Zhang, H.; Dixon, J.; Traphagen, N.; Wyatt, T.A.; Kharbanda, K.; Chadwick, S.S.; Kolliputi, N.; Allen-Gipson, D.S. Cigarette smoke impairs A2A adenosine receptor mediated wound repair through up-regulation of Duox-1 expression. Sci. Rep. 2017, 7, 44405.
- Sarr, D.; Gingerich, A.D.; Asthiwi, N.M.; Almutairi, F.; Sautto, G.A.; Ecker, J.; Nagy, T.; Kilgore, M.B.; Chandler, J.D.; Ross, T.M.; et al. Dual oxidase 1 promotes antiviral innate immunity. Proc. Natl. Acad. Sci. USA 2021, 118.
- Hewitt, R.; Farne, H.; Ritchie, A.; Luke, E.; Johnston, S.L.; Mallia, P. The role of viral infections in exacerbations of chronic obstructive pulmonary disease and asthma. Ther. Adv. Respir. Dis. 2016, 10, 158–174.