The renin–angiotensin system (RAS), an essential enzymatic cascade involved in maintaining blood pressure and electrolyte balance, is involved in the pathogenicity of COVID-19, since the angiotensin-converting enzyme II (ACE2) acts as the cellular receptor for SARS-CoV-2 in many human tissues and organs. In fact, the viral entrance promotes a downregulation of ACE2 followed by RAS balance dysregulation and an overactivation of the angiotensin II (Ang II)–angiotensin II type I receptor (AT1R) axis, which is characterized by a strong vasoconstriction and the induction of the profibrotic, proapoptotic and proinflammatory signalizations in the lungs and other organs. This mechanism features a massive cytokine storm, hypercoagulation, an acute respiratory distress syndrome (ARDS) and subsequent multiple organ damage.
1. Introduction
Coronaviruses are members of
Coronaviridae, a heterogeneous family of enveloped RNA viruses causing respiratory and enteric diseases in humans. Three potentially fatal diseases in humans caused by novel coronaviruses have been identified so far: the severe acute respiratory syndrome (SARS) in 2002, the Middle East respiratory syndrome (MERS) in April 2012, and lately, the coronavirus disease 2019 (COVID-19), caused by SARS-CoV, MERS-CoV and SARS-CoV-2, respectively [
1]. The novel coronavirus was shown to have similarity to its counterpart in bats, considered as the primary source of the virus, which could then have spread to humans either directly or from wild animals as intermediate hosts [
2,
3]. COVID-19 was first reported from Wuhan, China in December 2019, and spread worldwide two months later. Around 200 countries over the entire world have reported different numbers of cases. The disease has drastically expanded in the United States, Spain, Italy, Germany, France, China, Iran, the United Kingdom and Turkey [
1].
An array of clinical manifestations that vary in severity, from asymptomatic and very mild (fever and respiratory symptoms such as cough and shortness of breath) to severe (pneumonia, acute respiratory disease syndrome, or ARDS, and total organ failure) are associated with COVID-19 [
1]. Presently, over 250 million people worldwide have contracted COVID-19, and more than five million have died as a result of infection with SARS-CoV-2 [
1]. Elderly persons and those suffering from co-morbidities such as heart disease, lung disease, diabetes, hypertension and coronary artery disease (CAD) are at higher risk of developing severe COVID-19 illness. On 18 March 2020, a COVID-19 Response Team reported that 80% of COVID-19-related deaths were among the elderly, aged >65 years [
1,
4]. The main cause of death in COVID-19 patients is inflammation, especially in the lungs, which leads to ARDS [
5]. Unfortunately, no specific treatment is available for fatal and fast-spreading SARS-Cov-2 infection. However, a limited number of clinical studies have reported that some medications, such as antimalarial drugs (chloroquine and hydroxychloroquine), antiviral drugs (remdesivir) and dexamethasone (corticosteroid) have the potential to reduce the duration and symptoms of COVID-19 [
6]. Indeed, patients treated with chloroquine (CQ) recovered much sooner than other patients cured with other medicines [
7]. Furthermore, the treatment with hydroxychloroquine (HCQ) ameliorated the fever and decreased the cough duration [
8]. However, treatment with HCQ and CQ combined was not effective. A recent study by Hadjadj et al., found that patients with severe COVID-19 had impaired type-I interferon (IFN-I) activity, increased T cell apoptosis and inflammatory response, while the regulatory effect on the immune response by HCQ/CQ was not powerful enough to inhibit the over-activation of the innate immune system [
9]. Other findings suggested that SARS-CoV-2 can escape lysosome destruction by blocking the fusion between autophagosomes and lysosomes [
10,
11]. In addition, it has been reported that montelukast, a drug usually used in asthma, may be also proposed as an adjuvant in COVID-19 therapy due to its several beneficial properties (antiviral and antioxidant properties, prevention of neurological disorders due to SARS-CoV-2, limitation of the ischemia and the cytokine storm, improvement of respiratory symptoms) [
12] as well as dexamethasone, which could provide a beneficial effect in COVID-19 patients [
13].
SARS-CoV-2 single-stranded RNA genome encodes for the non-structural proteins (Nsps) and for four structural proteins: Nucleocapsid (N), Envelope (E), Membrane (M), and Spike (S), as well as the open-reading frame (ORF1a and ORF1ab) precursor polyproteins [
14]. The Nsp1 is considered to be a major virulence factor which blocks the translation of the messenger RNA through binding to the small ribosomal subunit, thus inhibiting the production of host proteins, including interferons. Therefore, Nsp1 favors viral replication by destroying the innate immunity defense mechanisms [
14]. However, the viral surface spike glycoprotein called “S-protein”, consisting of two domains (S1, S2) separated by a protease cleavage site [
3], is considered the key element of viral–host interaction by facilitating the virus’ entry into the hot cells [
3,
15]. In fact, the viral entry is mediated by the binding of the S1 domain to the host cell surface receptor “angiotensin converting enzyme 2 (ACE2)”, a major component and a key player of the Renin–Angiotensin system (RAS) [
15,
16]. The importance of ACE2 in the pathogenesis of SARS-CoV-2 infection has been of particular interest in recent months. The successful binding of the S1 receptor-binding domain (RBD) to the ACE2 peptidase domain (PD) will expose the S1/S2 inter-domain which, after cleavage by the host Transmembrane Serine Protease 2 (TMPRSS2), allows fusion of the viral and the host cell membranes, a mechanism mediated by the S2 domain [
3,
16]. Thus, SARS-CoV-2/RAS interaction is a key step favoring viral pathogenesis and COVID-19.
The RAS is a hormonal and physiological system whose function is to control blood pressure and circulating blood volume [
17]. The ubiquitous RAS, also referred to as the Renin–Angiotensin–Aldosterone system, thus plays an essential function in humans as being a regulator of pulmonary, cardiovascular, renal and innate immune functions and the gut microbiota [
18,
19]. In the RAS pathway, renin secreted by the kidney cleaves Angiotensinogen (AGT) secreted by the liver to give Angiotensin I (Ang I). The latter is cleaved by the angiotensin-converting enzyme (ACE) to produce Angiotensin II (Ang II), which is the substrate of ACE2 (
Figure 1). Ang II binds to the vasoconstriction-mediating type 1 (AT1R) and vasodilation-mediating type 2 (AT2R) receptors. Ang II can also be further cleaved by ACE2, into Angiotensin (1–7) [Ang-(1–7)], which interacts with the proto-oncogene G-protein-coupled receptor Mas (MasR) [
19,
20].
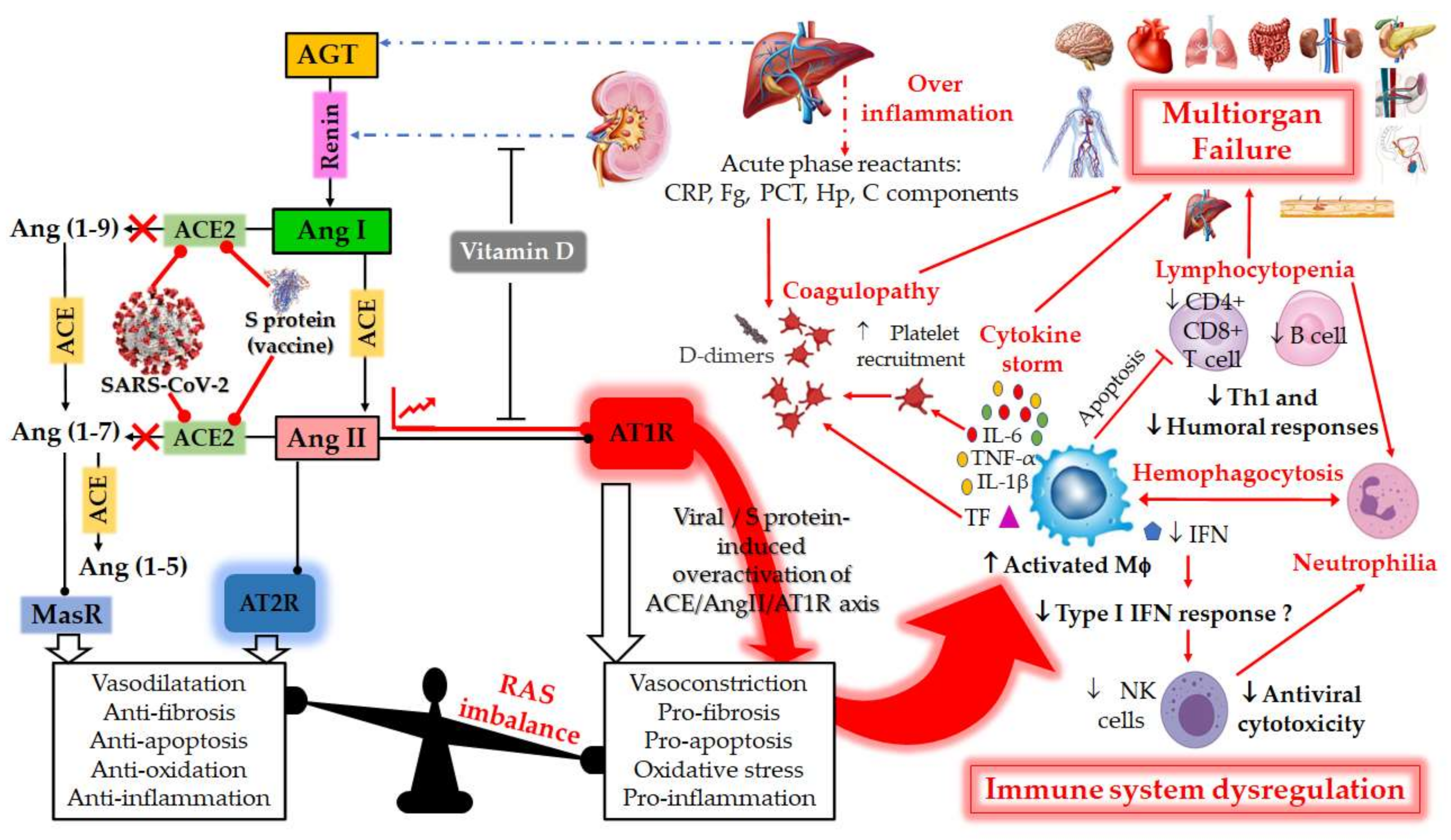 Figure 1.
Figure 1. Schematic diagram of the dysregulation in the Renin–Angiotensin System (RAS) and the host immune system caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) or by vaccination with mRNA encoding SARS-CoV-2 spike (S) glycoprotein. RAS is a metabolic cascade which supports a series of enzymatic reactions in which the liver secreted AGT is transformed into Ang I by renin, which is a protease secreted by juxtaglomerular kidney cells in response to decrease in blood pressure or sodium load in the distal convoluted tubule. Ang I is subsequently converted to Ang II by ACE which can bind to the AT1R to exert several actions, such as vasoconstriction, pro-fibrosis, pro-apoptosis, oxidative stress and pro-inflammation. ACE2 counterbalances Ang II/AT1R effects by cleaving Ang I and Ang II into Ang-(1–9) and Ang-(1–7), respectively. Ang-(1–9) is also converted into Ang-(1–7), a negative regulator of the RAS, which binds to the MAS receptor to exert protective actions of vasodilatation, anti-fibrosis, anti-apoptosis, anti-oxidative and anti-inflammation. Ang-II can also bind to AT2R to counteract the aforementioned effects mediated by AT1R. The balance between the Ang II/AT1R axis and the ACE2/Ang (1–7)/MasR axis is therefore maintained under physiological conditions. However, during SARS-CoV-2 infection or upon receiving a spike protein-based vaccine, the viral Spike (S) glycoprotein binding to ACE2 receptor induces overactivation of the ACE/Ang II/AT1R axis. This event prevents normal Ang II degradation, the excess of which leads to AT1R overactivation and RAS system imbalance. Such an imbalance is very deleterious for the human body, mainly due to the important immunomodulatory roles of ACE2, which can directly interact with macrophages in the setting of vascular and lung inflammation. Patients with severe COVID-19 infections show hallmarks of sepsis, widely explained by an exacerbation of macrophage activation, including excessive inflammation with the presence of acute phase reactants (such as D-dimer, CRP, etc.), impending cytokine storms and overexpression of IL-1β, IL-2, IL-6, and TNF-α in the early phase of the disease. These induce the production of a compelling number of factors linked to the coagulation cascade (TF, Fb, etc.) and resulting in the onset of thrombi and associated disseminated intravascular coagulation (DIC). The inflammatory response to SARS-CoV-2 also consists of lymphopenia occurring early in >80% of patients and is prognostic, manifested as reduction in—and functional exhaustion of—CD4+ more than CD8+ T cells. Such impaired T cell responses can result from deficient IFN production, as IFNs act on the antigen-presenting cells, T cells, and induce other cytokines and chemokines that regulate T-cell responses. These events lead to imbalance of the innate/acquired immune response, delayed viral clearance and unusual predominance of hyperstimulated macrophage and neutrophil in targeted injured tissues. The permanent immune activation in predisposed elderly adults and patients with cardiovascular risk can lead to hemophagocytosis-like syndrome, with uncontrolled amplification of cytokine production, leading to endothelial dysfunction, tissue damage and multiorgan failure, which is the starting point of a progression towards the serious and fatal complications of COVID-19. This syndrome results from the ineffective activation of cytotoxic CD8+ T lymphocytes and Natural Killer T lymphocytes, and leads to ineffective viral cytotoxicity and weak antibody production. NK cells are regulated by IFNs during coronavirus infection, and patients with severe COVID-19 showed profound depletion and functional exhaustion of NK cells, the dysfunction of which could be due to dysregulation of IFN responses. On the other side, Vitamin D could help avoid the potential deleterious COVID-19 effects sometimes observed following vaccination, by either inhibiting renin secretion or suppressing AT1R overactivation. AGT: angiotensinogen; Ang I: angiotensin I; ACE: angiotensin-converting enzyme; Ang II: angiotensin II; ACE-2: angiotensin-converting enzyme-2; Ang 1–7: angiotensin 1–7; AT1R: angiotensin II type 1 receptor; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; COVID-19: coronavirus disease 2019; CRP, c-reactive protein; IL-1β, interleukin-1β; IL-6, interleukin-6; TNF-α. Tumor Necrosis Factor-alpha; INF, Interferon; Fg, fibrinogen; PCT, procalcitonin; Hp, haptoglobin; C, complement; M
Φ, macrophage; NK, Natural Killer; Th1, T helper type 1; TF, tissue factor.
Recently, the dysregulation of RAS has been extensively investigated as being responsible for COVID-19 complex symptoms [
17]. During viral infection, SARS-CoV-2 over-activates the RAS by binding to the ACE2 receptor, which normally has the function of degrading Ang II [
21]. Thus, the binding of SARS-CoV-2 to the ACE2 receptor prevents the normal degradation of Ang II, the excess of which leads to over-activation of its cellular target, the AT1R. The over-activated AT1R is very deleterious for the human body, leading, in particular, to the appearance of COVID-19. The activated receptor induces vasoconstriction, hypertension, organ hypertrophy (heart, blood vessels), tissue fibrosis (heart, lungs, kidneys, and liver), ageusia (loss of taste), anosmia (loss of smell), neurological disorders, intestinal and vascular inflammation, obesity and action on glucose metabolism (diabetes), skin and testicular lesions [
12,
17,
20,
22] (
Figure 1). The location of ACE2 within the body is thought to be key to determining the progression of disease by viruses targeting this receptor, given the fact that SARS-CoV-2 relies on the binding of the viral S glycoprotein to ACE2 for viral entry into cells. Consequently, ACE2 would also be downregulated by SARS-CoV-2 in COVID-19 [
23]. Since the RAS has a central role in COVID-19 diseases, the selective targeting of its major components could be an essential treatment measure [
19]. Recently, there are suggestions to use AT1R antagonists as a strategy of increasing ACE2 levels for treating SARS-CoV-2 infections [
24,
25]. Treatments with AT1R antagonists and vitamin D supplementation ensure a simpler and effective way of improving ACE2 levels independently among COVID-19 patients. Moreover, Vitamin D has a negative regulatory role on the RAS [
19]. Suppression of RAS by AT1R antagonists results in a feedback-induced compensatory increase in renin. Vitamin D suppresses the compensatory increase in renin levels following the inhibition of the RAS by AT1R antagonists [
26].
2. RAS and COVID-19
2.1. RAS: A Portal Entry for SARS-CoV-2
The RAS is a metabolic cascade involved in the regulation of cardiovascular homoeostasis and cardiac remodeling [
27]. Besides regulating blood pressure and circulating blood volume as well as fluid and salt balance [
28,
29], this system also has a significant role in atherosclerosis pathogenesis and in endothelial dysfunction [
17,
30]. RAS major components are renin, angiotensin, AGT, ACE and AT1R [
31]. A homologue of ACE, named ACE2, with opposite effect, has been identified and considered to be a key player in the RAS [
32,
33]. ACE2 protein, a transmembrane metallo-carboxypeptidase of type I, is mainly found on the renal epithelium, endothelial cells of vasculature and Leydig cells of testes [
34]. Thus, ACE2 expression is ubiquitous, being also included in the lungs, gastrointestinal (GI) tract [
34] and the central nervous system, where it is involved in central regulation of the cardiovascular function [
27]. ACE2 is implicated in regulating blood pressure and the hemostasis of electrolytes and fluid, with functional influences on different organs such as blood vessels, heart, kidneys and eyes [
35]. Pathologically, ACE2 has also been associated with hypertension, stroke, dyslipidemia, cardiovascular and kidney diseases [
34].
2.2. RAS Imbalance and Underlying Pathologies
Since SARS-CoV-2 uses ACE2 as a key receptor for cellular entry, the latter subsequent downregulation will decrease Ang II degradation, leading therefore to its accumulation [
17,
33]. This contributes to a potential dysregulation and overactivation of RAS (
Figure 1) [
17,
47]. Studies have shown that RAS imbalance worsens COVID-19 prognosis and aggravates its pathogenesis [
17,
46]. Therefore, the most common underlying pathophysiology of COVID-19 is a viral acute respiratory distress syndrome (ARDS) coupled with the cytokine storm syndrome [
17].
According to current knowledge, the main cause of death in COVID-19 patients in intensive care unit (ICU) is the ARDS secondary to SARS-CoV-2 pneumonia [
35]. ARDS is characterized by a hypoxemia with increased capillary–alveolar permeability, reduced lung compliance, alveolar epithelial cell loss, neutrophil infiltration and a diffuse bilateral pulmonary infiltrate that could lead to alveolar and interstitial remodeling and fibrosis [
48]. This could lead to the loss of pulmonary perfusion regulation and hypoxic vasoconstriction, as well as a low ventilation perfusion ratio [
2], thus requiring mechanical ventilation [
48]. According to Richardson et al., 80% of patients who required mechanical ventilation after COVID-19 infection evolved to death, emphasizing that ARDS is an underlying pathophysiology in COVID-19 patients which may be responsible for the high mortality rates [
49]. It has been shown that RAS imbalance may influence the pathogenesis of ARDS through Ang II and bradykinin [
48]. Experiments have revealed that rats with knock-out ACE2 exposed to non-SARS lung damage (such as endotoxin) developed a severe ARDS compared to the wild type rats [
50]. Therefore, ACE2 was shown to have a protective effect in rat models of acute lung injury, with ACE, Ang II and AT1R being considered as lung injury-promoting elements [
50]. Imai et al., have shown an upregulation of Ang II by ACE in the pathogenesis of acute lung injury through the AT1a receptor, leading therefore to severe lung failure [
28]. A study conducted by Kuba et al., showed that the blockage of ACE2 or its genetic manipulation leads to increased lung edema, vascular permeability and neutrophil accumulation [
50].
2.3. RAS Component Polymorphism
Many studies described the association of the RAS component genetic variation with the prevalence of COVID-19 diseases [
47,
68]. Therefore, it has been thought that genetic factors may render the host resistant or susceptible to infection with SARS-CoV-2 [
47]. The prevalence and disease outcome were linked to
ACE polymorphisms [
36]. The genetic polymorphisms of
ACE1 and
ACE2 genes may change their levels of expression, therefore leading to an increase in capillary permeability, coagulation, fibrosis and apoptosis in alveolar cells [
69]. In a pilot study conducted by Cafiero et al., it was found that some genetic variants in the RAS pathway may be potential actors for determining the clinical outcome and the pathological conditions associated COVID-19, such as DIC, interstitial pneumonia, thrombosis, conjunctivitis and the cytokine storm [
35]. Thus, inflammation and lung injury caused by ACE2 decrease, following viral binding, could be negatively affected by
ACE’s different genotypes, which in turn could increase ACE expression levels and then those of Ang II [
36]. The knowledge of these polymorphisms could help the management of COVID-19 infected patients [
36]. The major RAS component polymorphisms are illustrated in
Table 1.
Table 1. Human gene mutations and polymorphisms of the Renin–Angiotensin System associated with COVID-19.
RAS
Component Gene |
Chromosomal Location |
Associated
Disease/Phenotype |
Mutations,
Polymorphisms and
rs Number |
Allele/Genotype
Frequencies in Populations and Ethnicities |
References |
| ACE1 |
17q23.3 |
CVD
Kidney disease
Autoimmune diseases
Hypertension
Hypercoagulability
ARDS
Type 2 diabetes
Risk of obesity |
Insertion/Deletion (I/D) of a 287-bp Alu repeat in intron 16
(rs1799752) |
Lebanese
I: 0.27, D: 0.73 |
[77] |
Indians
I: 0.55, D: 0.45
Whites
I: 0.5, D: 0.5
African Americans
I: 0.41, D: 0.59 |
[76] |
British
I: 0.31, D: 0.69 (ARDS)
I: 0.49, D: 0.51 (healthy population) |
[73] |
Chinese
I: 0.705, D: 0.295 |
[75] |
Italians
I: 0.342, D: 0.658 |
[56] |
Italians
I: 0.27, D: 0.73 |
[35] |
Germans
I/I: 0.27, I/D: 0.43, D/D: 30 |
[48] |
Indians
I: 0.575, D: 0.425 |
[78] |
| ACE2 |
Xp22.2 |
Cardiovascular risk, Retinopathy in type-2 Diabetes Mellitus, Hypertension and Hypertensive left ventricular hypertrophy |
c.*1860-449C > T
SNP (rs2074192) |
Italians
C: 0.56, T: 0.44 |
[35] |
c.*264+788T > C
(rs2106809) |
Italians
A: 0.77, G: 0.33 |
c.2115-268A > T
SNP (rs233574) |
Africans
C: 0.92, T: 0.08
Europeans
C: 0.67, T: 0.33
East Asians
C: 0.996, T: 0.004
South Asians
C: 0.814, T: 0.814
Americans
C: 0.767, T: 0.233 |
[79] |
c.1402A > G
p.Ile468Val
SNP (rs191860450) |
East Asians with an allele frequency (AF) = 0.011 |
[16] |
c.1022A > G
p.Lys341Arg
SNP (rs138390800) |
Africans
AF = 4 × 10−3 |
c.2191C > T
p.Leu731Phe
SNP (rs147311723) |
Africans
AF = 0.014 |
c.631G > A
p.Gly211Arg
SNP (rs148771870) |
Europeans AF = 2 × 10−3
South Asians AF = 1.9 × 10−3 |
c.2089A > G
p.Arg697Gly
SNP (rs751603885) |
South Asians
AF = 2.4 × 10−3 |
c.2074T > C
p.Ser692Pro
SNP (rs14903946) |
Africans
AF = 6 × 10−3 |
c.55T > C
p.Ser19Pro
SNP (rs73635825) |
Africans
AF = 3 × 10−3 |
| AGT |
1q42.2
1q42–43 |
Hypertension
Heart failure Myocardial infraction |
c.704T > C
p.Met235Thr
(aka Met268Thr)
SNP (rs699) |
Tunisians
M/M: 0.291, M/T: 0.291
T/T:0.419 |
[80] |
Vietnamese
T: 0.92, M: 0.08 |
[81] |
Iranians
T: 0.39, M: 0.61 |
[82] |
Indians
M: 0.52, D: 0.48 |
[78] |
New Zealanders
T/T: 0.19, T/M: 0.47, M/M: 0.34 |
[83] |
c.521C > T
p.Thr174Met
SNP (rs4762) |
New Zealanders
T/T: 0.7, T/M: 0.2, M/M: 0.1 |
[83] |
| AT1R |
3q21–q25 |
Systolic blood pressure
Left ventricular hypertrophy Hypertension
Aortic stiffness Myocardial infarction
Carotid intimal-medial thickening, CAD and stroke, Overweight, Diabetes |
c.1166A > C
in the 3′ UTR
SNP (rs5186) |
Egyptians
C:0.24, A:0.76 (control group)
C:0.34, G:0.66 (premature CAD patients) |
[84] |
Jordanians
Higher frequency of A allele |
[85] |
Iranians
Higher frequency of A allele |
[86] |
| AT2R |
Xq23–26 |
Metabolic syndrome |
−1332A > G
SNP (rs1403543) |
Egyptians
A:0.55, G:0.45 (control group)
A:0.41, G:0.50 (premature CAD patients) |
[84] |
3. Correlation of Habits, Gender and COVID-19 with RAS Polymorphism
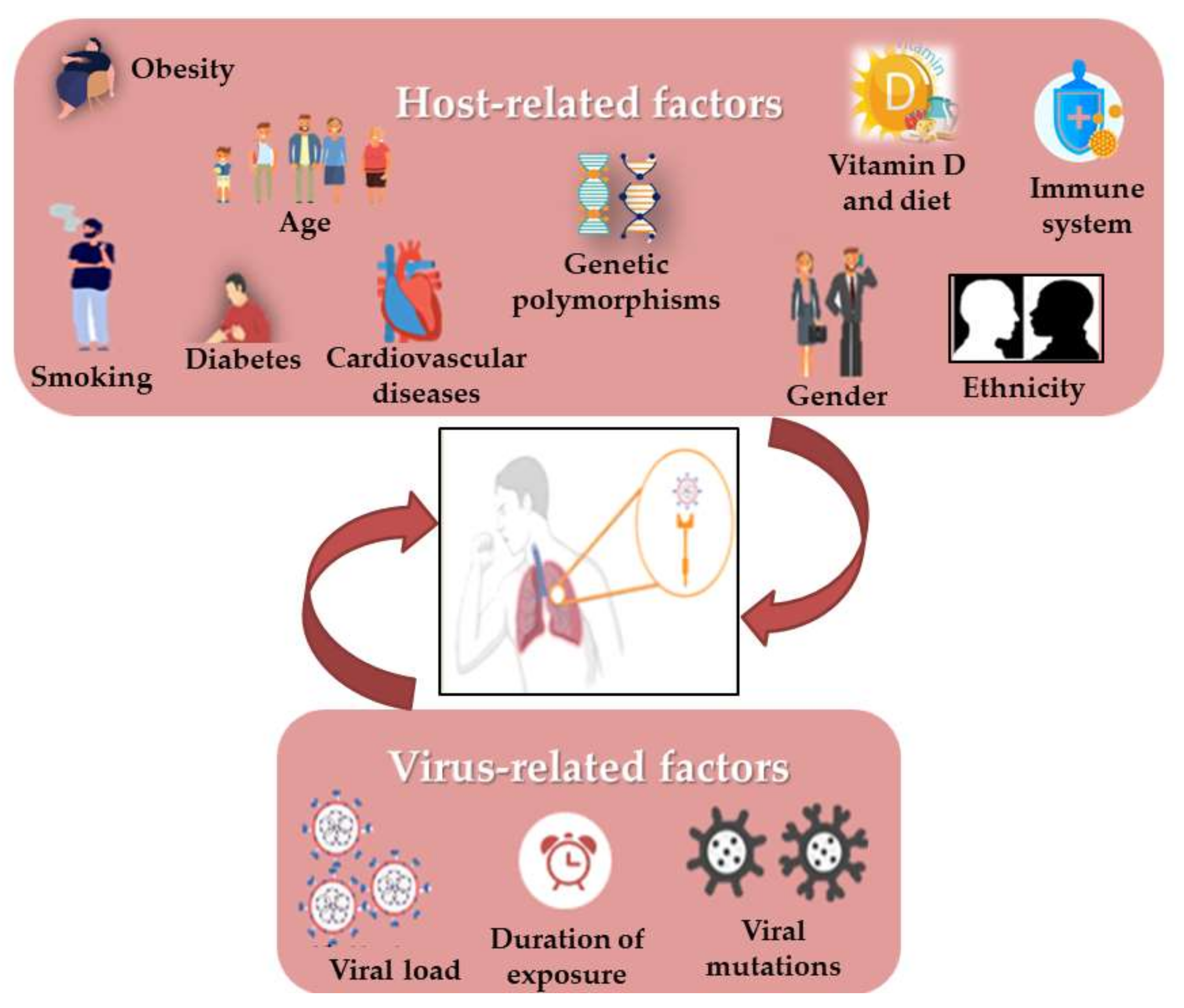
Still now, it is surprising that some COVID-19 patients are asymptomatic while others have much more severe disease outcomes. Moreover, viral respiratory infections are generally more harmful in children than in adults, but this appeared to be inverted in SARS-CoV-2 infection. It has been shown that some virus-related factors (viral load in the inoculum, the exposure duration and viral genomic mutations) can influence the severity and outcome of the disease [
100] (
Figure 2). Similarly, it quickly became obvious that several risk factors such as age, gender, the presence of comorbidities (such as smoking, immune status, diabetes, cardiovascular disease including hypertension, respiratory diseases and cancer) and the genetic background seem to control the manifestations and outcome of infection [
41,
45,
101,
102]. In addition, it has also drawn attention to vitamin D deficiency [
103], as well as the ethnic differences, such as the black and South Asian ethnicities, and the lower socioeconomic statuses which are considered to increase risks [
54]. Most mortalities occurred in the elderly and in men compared to women (4.7% vs. 2.8%). Moreover, the death of patients with no pre-existing conditions is approximately 10 times lower than in those with pre-existing ones. As in the 2003 SARS epidemic, hypertension has the most comorbidity frequency in non-survivors, in addition to cardiovascular diseases, obesity and diabetes, especially in smokers [
54,
90]. There is evidence that the RAS upregulation in the adipose tissue may lead to hypertension and insulin resistance in obese people. [
37]
4. Conclusions
The SARS-CoV-2 outbreak has threatened several sectors in the world, including the economic and public health structure, and has furthermore caused hundreds of thousands of confirmed deaths, observed especially in elderly and immunocompromised people. Many studies are currently ongoing to understand the molecular mechanisms of COVID-19 cellular invasion, and thus the cascade activation and/or suppressing action detected among host cells. The normal signaling pathway of the RAS is clearly disturbed during viral infection due to the downregulation of ACE2 and the activation of Ang II/AT1R axis, which favors a pro-inflammatory state leading to cytokine storm, immunothrombosis, ARDS and multiple organ damage in COVID-19 patients. In addition, the manifestations and poor outcome of infection seem to be controlled by several risk factors, the presence of comorbidities and the genetic background of each individual. As a treatment, some brands worked hard to design a new vaccine that would protect the population and decrease the disease’s severity after taking two or more doses. More than 5.85 billion vaccines have been administrated now across 184 countries, according to some data. However, knowing that COVID-19 regularly enters into a mutation cycle, more efforts should be made on more efficient treatments to defeat SARS-CoV-2 definitely. Therefore, targeting the RAS could be an important therapeutic challenge in order to try to restore its equilibrium. In other words, several possibilities could be pointed out for future treatment of COVID-19, including (1) the use of stimulators or agonists of the protective pathway components to favor their function, such as an ACE2 protein-based vaccine or a recombinant ACE2, AT2R or MasR stimulator, or (2) the use of inhibitors targeting the Ang II/AT1R axis, such as AT1R blockers or Ang II antagonists. Therefore, these promising RAS therapeutic treatments aiming at reducing or even preventing COVID-19 diseases should be well studied in order to be supported by valuable conclusive data for their definitive usage.
This entry is adapted from the peer-reviewed paper 10.3390/molecules26226945