This review deals with the role of the secreted enzyme, autotaxin (ATX), in the progression of breast cancer. ATX produces lysophosphatidate (LPA), which signals through six G-protein coupled receptors, promoting tumor growth, metastasis, immune evasion and survival from chemotherapy and radiotherapy. Many cancer cells produce ATX, but breast cancer cells express little ATX. Instead, in breast cancer, ATX is produced by tumor-associated stroma. Breast tumors are also surrounded by adipose tissue, which is a major bodily source of ATX. In mice, a high-fat human type diet increases ATX production in adipocytes. ATX production in obesity is also increased because of low-level inflammation in the expanded adipose tissue. This increased ATX secretion and consequent LPA signaling is associated with decreased adiponectin production, which results in adverse metabolic profiles and glucose homeostasis. Increased ATX production by inflamed adipose tissue could contribute to the association between obesity and breast cancer. This would result from the cross talk between breast tumors and adjacent adipose tissue. Breast tumors produce inflammatory mediators that stimulate ATX transcription in adipocytes adjacent to the tumors. This drives a feedforward inflammatory cycle since increased LPA signaling increases the production of more inflammatory cytokines/chemokines and cyclooxygenase-2 resulting in more ATX secretion. This cycle is typical of a wound healing response, which in the case of cancers become maladaptive. Thus, inhibiting ATX activity derived from adipocytes and/or tumor stromal cells has implications as an adjuvant for breast cancer treatments by attenuating the inflammatory cycle. Targeting ATX activity and LPA signaling could potentially increase the efficacy of chemotherapy and radiotherapy independently of the breast cancer type because most ATX is not derived from breast cancer cells. Blocking ATX activity and LPA signaling could also decrease morbidity from radiation-induced fibrosis.
Dear author, the following contents are excerpts from your papers. They are editable.
(Due to the lack of relevant professional knowledge, our editors cannot complete a complete entry by summarizing your paper, so if you are interested in this work. you may need to write some contents by yourself. A good entry will better present your ideas, research and results to other scholars. Readers will also be able to access your paper directly through entries.)
1. Autotaxin and LPA Metabolism
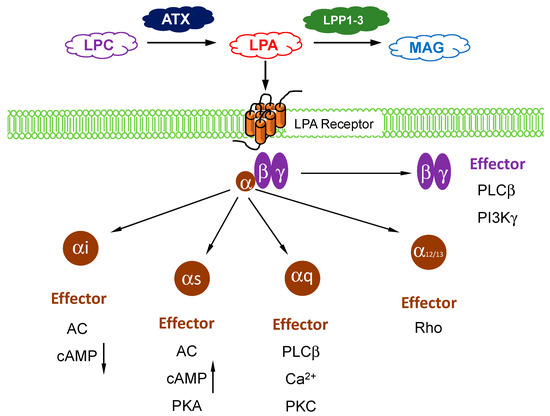
Autotaxin (ATX) belongs to a family of ectonucleotide pyrophosphatase/phosphodiesterases (ENPPs) and its gene name is
ENPP2 [
1]. There are five other members of this family and these hydrolyze phosphodiester bonds in nucleotide phosphates [
2]. By contrast, secreted ATX acts primarily as a lysophospholipase D, which converts extracellular lysophosphatidylcholine (LPC) into lysophosphatidate (LPA). The affinity of ATX for LPC is ~10-fold higher than for nucleotide substrates [
3]. ATX was discovered in culture medium from melanoma cells because of its effects in stimulating cell migration [
4]. It was not until a decade later that this cell migration effect was shown to depend on its production of lysophosphatidate (LPA) [
5,
6]. In fact, most of the biological functions of ATX are attributed to signaling by LPA [
7]. ATX acts as the “gatekeeper” to control LPA signaling through a family of six G protein-coupled receptors (). The LPA receptors are widely expressed in different cells and they regulate a wide range of signaling pathways through their coupling to Gi, Gs, Gq, and G12/13 () [
8,
9].
Figure 1. Overview of lysophosphatidate (LPA) signaling pathway. Extracellular LPA is produced from the enzymatic action of autotaxin (ATX) on lysophosphatidylcholine (LPC). LPA is degraded by lipid phosphate phosphatases (LPP)1–3 into inactive monoacylglycerol (MAG). LPA signals through at least six known G-protein coupled receptors (with three sub-units) to mediate its downstream cellular effects, which are dependents on the coupling and/or subunit type.
2. Role of ATX Activity in Wound Healing, Chronic Inflammation, and Cancer
One of the important functions of ATX and LPA signaling is in wound healing. ATX is secreted by activated platelets and secretion from tissues is increased by inflammation that is caused by tissue damage or infections [
29,
30]. The consequent increase in LPA signaling stimulates wound repair by activating the migration and division of fibroblasts and keratinocytes in the wounded area [
31] and by facilitating collagen deposition [
32], which leads to the formation of scar tissue. LPA also stimulates angiogenesis by activating the migration and proliferation of vascular endothelial cells [
33,
34] to provide the new blood vessels necessary for tissue repair. LPA also induces lymphocyte homing [
35] and transformation of monocytes to macrophages [
36], which is an important part of the host defense system. When the wound is healed, inflammation resolves and the secretion of ATX falls back to basal conditions [
29,
37].
There are many conditions in which inflammation is not resolved. These chronic inflammatory conditions include pulmonary fibrosis, cirrhosis, obesity, rheumatoid arthritis, inflammatory bowel disease, cardiovascular diseases, and cancers [
8,
38]. In fact, the wound healing functions of ATX and LPA are hijacked in cancers (wounds that do not heal) [
39,
40,
41,
42]. Inflammation and decreased acquired immune responses are “hallmarks” of cancer [
8,
41,
43]. Chronic LPA signaling enables cancer cells to evade the immune system [
44,
45,
46] and the activation of LPA5 receptors suppresses the function of CD8+ cytotoxic T cells by inhibiting the mobilization of intracellular Ca
2+ and extracellular signal-related kinases (ERK) activation [
47]. LPA when bound to low-density lipoproteins (LPL) promotes platelet activity, which leads to stress fiber production and vascular endothelial damage. Mechanistically, LPA signaling through TLR4 receptors, which is a toll-like receptor partly responsible for innate immune system activation, upregulates nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) activation. This may contribute to the creation of a chronic inflammatory milieu central to the progression of atherosclerosis [
48,
49]. LPA also increases vascular endothelial growth factor (VEGF) production, which stimulates the angiogenesis needed for tumor growth [
50]. LPA signaling is generally increased in cancers because of the high secretion of ATX and the low expressions of LPP1 and LPP3 [
51]. ATX concentrations are correlated with invasiveness [
8,
10,
52] and the ATX gene (
ENPP2) is among the 40–50 most up-regulated genes in metastatic tumors [
53,
54,
55].
The imbalance in expression between macrophage types M1 and M2 is implicated in the development of auto-immunity and obesity in murine models [
56]. Typically, M1 macrophages are labeled as classically activated or inflammatory macrophages and M2 macrophages as alternatively activated, or wound-healing. Normally, macrophages retain their imprinted phenotype but they have sufficient plasticity to be remodeled as needed in response to acute inflammatory cytokines, which increases the M1/M2 ratio [
57]. Adiponectin contributes toward an anti-inflammatory profile with increased M1 macrophage expression in adiponectin knockout mice, and blocks M1 marker expression in human circulating monocyte-derived macrophages and stromal vascular fraction cells in human subcutaneous fat [
58].
Recently, it was reported that tumor-associated macrophages may be the predominant source of LPA production in the ascites of ovarian cancer patients, and that CD163+CD206+ tumor-associated macrophages play an essential role as the main producers of ATX and phospholipase A2s (PLA2s) [
59]. While tumor cells express predominantly LPA1–3 receptors, macrophages and T cells selectively express LPA5 and LPA6 receptors at high levels, which points to cell type-selective LPA signaling pathways in a cancer microenvironment [
42,
60]. Antagonizing the actions of the LPA5 receptor has been proposed as an essential target for the immunological control of cancer progression [
60].
3. Role of Adipose Tissue as a Source of ATX Production in Breast Cancer
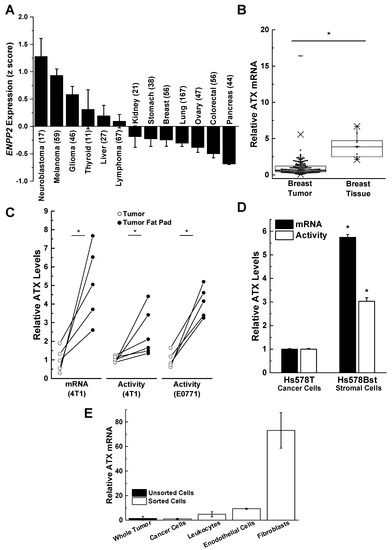
ATX is secreted directly in highly significant amounts by melanoma, glioblastoma, glioma, and thyroid tumors when compared to stomach, breast, lung, ovary, colorectal, and pancreatic tumors [
61,
62,
63,
64] (A). ATX mRNA levels and ATX activity are relatively low in human breast tumors compared to adjacent breast tissue rich in adipose tissue (B). A similarly low ATX activity is seen in mouse 4T1 and E0771 breast tumors compared to adjacent adipose tissue [
7,
62,
65,
66,
67] (C). This relationship is also illustrated by the comparison of human Hs578T breast cancer cells relative to Hs57Bst stromal cells isolated from the same tumor (D). When the cells from mouse 4T1 breast tumors were separated after collagenase digestion, the majority of the ATX mRNA was associated with the fibroblast fraction rather than with endothelial cells or leukocytes. The breast cancer cells had very low ATX mRNA expression (E). These experiments on ATX mRNA expression provide information on where ATX is produced before it is secreted into the tumor microenvironment where it attaches to adjacent cells including cancer cells by binding to integrins and syndecan-4. This binding appears to selectively channel LPA signaling to LPA receptors [
68,
69,
70,
71]. It is, therefore, concluded that most of the ATX that mediates LPA signaling is produced by adipocytes, fibroblasts, and tumor-associated stromal cells rather than the breast cancer cells themselves.
Figure 2. Breast cancer cells are poor producers of autotaxin (ATX) compared to adjacent adipose tissue tumor-associated fibroblasts. (
A) Human breast cancer cells express little ATX compared to other neuroblastoma, melanoma, glioma, thyroid, and liver cancer cells. Results are means ± SEM. Numbers in parentheses indicates the number of cell lines. Results taken from cBioPortal (
www.cbiportal.org) [
63,
64] and are reproduced from Reference [
29] with permission. (
B) ATX mRNA expression in 176 human breast tumors and 10 normal breast tissue specimens. Box plots show minimum, mean, and maximum values, 25th, 50th, and 75th percentiles (box), and 1st and 99th percentiles. Results are expressed relative to the mean of the breast tumor results, which were given the value of 1. *
p < 0.001. Adapted from Reference [
67]. (
C) ATX, mRNA, and activity levels are significantly lower in tumors compared to adjacent fat pads in orthotopic syngeneic and immunocompetent mouse models (4T1/BALB/C, E0771/C57BL/6) *
p < 0.05 by a paired
t-test. Results are expressed relative to the mean of the breast tumor results, which were given the value of 1. Includes results adapted from Reference [
14]. (
D) Relative ATX mRNA and activity levels in patient-matched Hs578T breast cancer cells and Hs578Bst stromal cells. Results are means ± SEM from three independent experiments. *
p < 0.05 vs. Hs578T breast cancer cells. Adapted from Reference [
67]. (
E) ATX expression in mouse 4T1 tumors comes predominantly from cancer-associated fibroblasts. Whole 4T1 tumors were enzymatically digested and sorted by flow cytometry for cancer cells (epithelial cells) using EPCAM (epithelial cell adhesion molecule), leukocytes using CD-45, endothelial cells using CD-31, and cancer-associated fibroblasts using platelet-derived growth factor alpha (PDGFα). ATX mRNA levels are expressed relative to those in the whole tumor. Results are means ± SEM from three independent experiments for whole tumor and cancer cells, and means ± range for two independent experiments for leukocytes, endothelial cells, and fibroblasts.
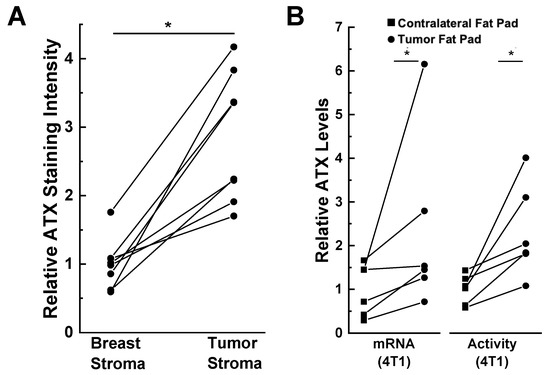
The presence of the tumor also influences this expression of ATX. This is illustrated by immunostaining of human tissues where ATX is present at higher concentrations in human breast tumor stroma compared to the adjacent breast stroma (A) [
67]. Furthermore, ATX mRNA expression and activity in the fat pad adjacent to 4T1 breast tumors in mice is higher than in the contralateral fat pad that did not contain a tumor (B) [
14]. Popnikolov et al. [
72] also used immunostaining for ATX and showed positivity for ductal carcinomas. There was strong ATX staining in peritumoral fibroblasts, whereas the cancer cells were weakly positive. In addition, ATX staining was low in normal ducts and lobules compared to the carcinomas. It is, therefore, important to consider where ATX is produced and where the secreted protein is expressed. ATX production in breast cancer cells and normal epithelial cells is low compared to that in breast adipocytes and fibroblasts.
Figure 3. ATX is induced in tumor-associated compared to normal breast adipose tissue. (
A) ATX immunohistochemical staining is increased in human tumor stroma compared to adjacent breast stroma. *
p < 0.001 by paired
t-test. Staining intensity was quantified by ImageJ [(NIH), Bethesda, MD, USA] and the results were adapted from Reference [
67]. (
B) ATX mRNA and activity levels are higher in tumor-bearing mammary fat pads compared to unaffected contralateral fat pads in a 4T1 orthotopic, synergistic, and immunocompetent BALB/C mouse model. ATX staining and mRNA levels are expressed relative to the breast stroma and contralateral fat pad, respectively. *
p < 0.05 by paired
t-test. Results adapted from Reference [
14].
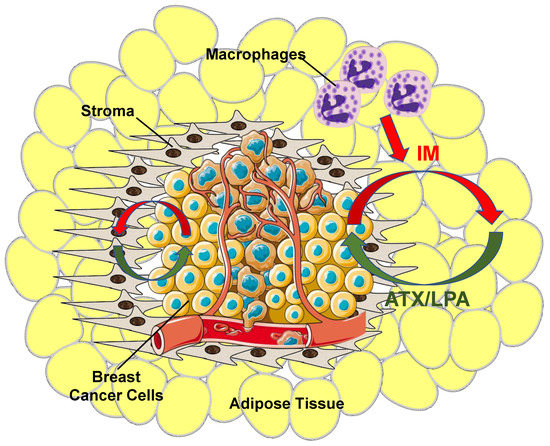
It was concluded from this combined work that adipocytes and fibroblasts in the proximity of the cancer cells are stimulated by inflammatory cytokines produced by the tumors and that this increases ATX synthesis and secretion [
9,
23,
67] (). This amplifies the inflammatory cycle and promotes accumulation of inflammatory macrophages [
44,
45]. This evidence for bi-directional signaling between breast tumors and adipose tissue through the ATX-LPA-inflammatory cycle has since been confirmed by others [
73,
74].
Figure 4. Overview of ATX/LPA signaling within the breast tumor microenvironment. Breast cancer cells produce virtually no ATX relative to tumor stroma and surrounding adipose tissue. Instead, as cancer cells grow, they establish an inflammatory milieu where inflammatory mediators (IM) (red arrows) stimulate both tumor stroma cells, including tumor-associated fibroblasts, and adjacent adipose tissue to increase ATX production. The tumor also recruits other circulating cells including macrophages to further increase inflammatory signaling and promote a pro-survival and pro-growth environment. Increased ATX enzymatic activity increases tumor LPA concentrations (green arrows), which, thereby, initiates a vicious cycle that further fuels tumor growth and ultimately metastasis.
4. Role of Lipid Phosphate Phosphatases in Controlling LPA Signaling and Tumor Progression
Another important component in the regulation of LPA signaling in cancers is the degradation of LPA by LPP1 and LPP3 (
PLPP1 and
PLPP3, respectively). The expressions of these two LPPs are decreased in lung, ovarian, and breast tumors [
97,
98,
99] and this can contribute to the increase of LPA concentrations in the tumors [
100,
101,
102]. Low expression of mRNA for LPP1 is one of 12 changes in mRNA that predicts poor survival in breast cancer patients [
103]. We showed that mRNA concentrations for LPP1 were lower in all types of breast tumors compared to normal breast tissue [
27,
51].
The consequence of low LPP1 activity has a two-fold implication. First, there is less degradation of LPA in the vicinity of the tumors. Second, expression of LPP1 attenuates signaling downstream of LPA and protease activated receptors and this is not related to the dephosphorylation of extracellular LPA. The LPPs are expressed on internal membranes as well as on plasma membranes where they dephosphorylate bioactive lipid phosphates and pyrophosphates including phosphatidate and ceramide 1-phosphate, which are involved in intracellular signaling [
21,
104,
105]. This could explain why increasing LPP1 activity attenuates the activation of Ca
2+-transients [
106] and phosphatidate accumulation after phospholipase D activation [
107]. These presumed intracellular actions are downstream of receptor activation because LPP1 attenuates the effects of an LPA analogue (wls-31) that activates LPA receptors, but cannot be dephosphorylated [
106,
107]. Furthermore, LPP1 attenuates signaling by the protease-activated receptor-1 (PAR1) in MDA-MB-231 breast cancer cells [
106]. This latter effect of LPP1 requires phosphatase activity and it cannot be explained by dephosphorylation of extracellular LPA. Thrombin-induced ERK phosphorylation is also inhibited by LPP1 [
108]. One interpretation of these results is that the LPPs dephosphorylate a lipid mediator that facilitates signaling downstream of the activation of several different types of GPCRs [
21,
104]. Thus, the low activity of LPP1 in cancer cells makes them hypersensitive to the effects of ATX and signaling by LPA [
10].
Increasing the low levels of LPP1 in breast cancer cells decreases cell division and blocks tumor growth and metastasis in a mouse breast cancer model by up to 80% [
27,
106]. The low expression of LPP1 in cancer cells is associated with increased expression of the metalloproteinases, (MMP)-1, 3, 7, 9, 10, 12, 13 and cyclin D1/D3, which are transcribed downstream of the AP1 (Fos-Jun) complex and the tumors have increased expression of c-Fos and c-Jun [
27]. The increased expression of MMPs is associated with decreased collagen content in the tumors of experimental mice. This low collagen content could allow cancer cells to exit from the breast tumor, enter the circulation, and metastasize to other organs. Breast tumors from patients also show increased expression of MMP-1, 7, 8, 9, 12, 13 and the tumors have increased expression of c-Fos and c-Jun. This could contribute to increased metastasis and explain why breast cancer patients that have low expression of LPP1 in their tumors exhibit increased mortality [
27]. This relationship was not statistically significant for LPP3 in breast cancer patients [
27] even though increasing LPP3 expression in mouse ovarian cancer cells attenuates their ability to support tumor growth [
106,
109].
In contrast to this, LPP2 has a completely different effect and mRNA concentrations for LPP2 (PLPP2) are increased in breast, lung, and ovarian tumors [
21]. A genomic screen between normal and transformed mesenchymal stem cells showed that LPP2 is elevated in several cancer cell lines including MCF7, SK-LMS1, MG63, and U2OS cells [
110]. We showed that increasing LPP2 expression in fibroblasts stimulates the cell division [
111]. Increased LPP2 expression in cancer cells is part of the transformed phenotype and it facilitates anchorage-dependent cell growth [
110]. Our unpublished work shows that knockout of LPP2 in MDA-MB-231 breast cancer cells decreases tumor growth by ~70% in a mouse breast cancer model. Although increased LPP2 activity increases the degradation of extracellular LPA, which would decrease tumor progression, the selective effects of LPP2 on intracellular signaling likely account for its negative effects on tumor growth.
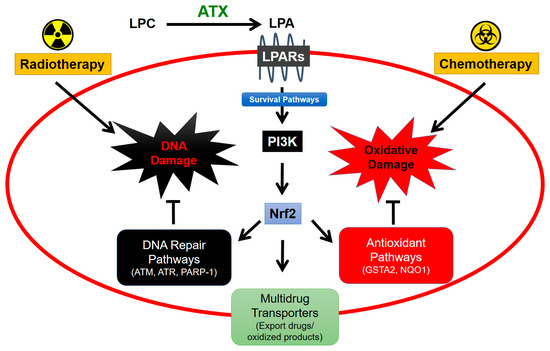
5. Role of Adipose Tissue-Derived ATX in Responses of Breast Tumors to Chemotherapy
Adipose tissue plays an important role in breast cancer and its treatment because adipocytes are the major source of ATX activity in the breast. The secretion of ATX is increased further by the inflammatory signals that the adipocytes receive from cytokines/chemokines secreted by cancer cells, stromal cells, and leukocytes. The survival signals generated by ATX through subsequent signaling through LPA receptors on tumor-associated cells decrease the efficacies of pacitaxel [
65], tamoxifen [
112], and doxorubicin [
113] in killing the breast cancer cells. These are major therapeutics for different types of breast cancer. Axiomatically, inhibiting ATX activity increased the efficacy of doxorubicin in decreasing breast tumor growth and metastasis in mice [
114]. This action is indirect and occurs because of decreased LPA production and activation of LPA1 receptors and phosphoinositide 3-kinase (PI3K). This signaling stabilizes the transcription factor, nuclear factor erythroid-derived 2-like 2 (Nrf2), which activates the anti-oxidant response element, and leads to the synthesis of anti-oxidant proteins and multi-drug resistance transporters [
113] (). These changes protect cancer cells by decreasing oxidative damage and by exporting chemotherapeutic drugs and toxic oxidation products.
Figure 5. ATX signaling protects cancers cells from cytotoxic effects of radiotherapy and chemotherapy. Lysophosphatidate (LPA) signaling stabilizes Nrf2 expression via PI3K-mediated survival pathways [
115]. Nrf2 facilitates expression of proteins involved in DNA repair and antioxidant pathways as well as increases expression of multidrug-resistant transporters on the cancer cell surface for export of drug and oxidized molecules from the cell [
29]. Combined, these mechanisms contribute to cancer cell survival and resistance to cancer therapy.
6. Role of Adipose Tissue-Derived ATX in Responses to Radiotherapy for Breast Cancer
In the treatment of breast cancer, ~60% of patients have their tumors removed surgically (lumpectomy). This is followed by radiotherapy (RT) to the post-surgical breast with daily fractions of 1.8–2 Gy/fraction to a total dose of 45–50 Gy. The most recent American Society for Radiation Oncology (ASTRO) guidelines for women with invasive breast cancer recommend hypo-fractionated RT with either 40 Gy in 15 fractions or 42.5 Gy in 16 fractions, i.e., ~2.6 Gy per fraction [
116]. This creates a fairly unique situation in which breast adipose tissue is irradiated and damaged multiple times.
We, therefore, investigated the effects of radiation-induced damage to adipose tissue. Exposure of cultured human breast adipose tissue to 0.5 to 5 Gy of γ-radiation increased the production of ATX as well as the LPA signaling through increased expression of LPA1 and LPA2 receptors downstream of DNA damage [
117]. These events caused activation of a feed-forward inflammatory cycle including an increase in the expression of COX-2 and multiple inflammatory cytokines/chemokines, which, in turn, increase more ATX secretion [
44,
45]. The inflammatory cycle could be attenuated by inhibiting the RT-induced activation of ATM, poly [ADP-ribose] polymerase-1 (PARP-1), and NFκB.
This work in vitro was extended in vivo using precision RT on a mammary fat pad in mice with a small-animal “image-guided” RT platform (SARRP) with integrated computed tomography (CT)-imaging. This system allows for treatment-planned RT to the tumor and fat pad while minimizing peripheral tissue damage. In these studies, a single dose of RT increased plasma ATX concentrations [
118]. There was no significant effect of one dose of RT on plasma concentrations of IL-6 and TNF-α, but three fractions of RT substantially increased these inflammatory cytokines [
118]. A similar increase was observed after three fractions of RT for VEGF, G-CSF, CCL11, and CXCL10 in the irradiated adipose tissue [
118]. These effects of multiple fractions of RT likely depend on the cumulative DNA and tissue damage. In addition, repeated doses of RT increase Nrf2 expression [
118], which raises the synthesis of numerous proteins that attenuate oxidative damage and promote DNA repair [
119,
120] (). Previous work predicted that Nrf2 blockade could provide a target for increasing the efficacy of RT by attenuating DNA repair [
121]. The results indicate that RT-induced expression of ATX appears to be an early event in vivo following RT-induced damage and that subsequent LPA signaling could then augment the release of inflammatory cytokines and chemokines to produce an inflammatory response.
LPA also decreases adiponectin secretion [
89]. Secretion of adiponectin by adipose tissue normally produces an anti-inflammatory response and it is inversely linked to the risk of obesity-associated malignancies and insulin resistance [
122]. Plasma adiponectin levels were decreased significantly by three fractions of RT in non-tumor bearing mice [
118] and this could also be a response to increased LPA signaling. Plasma adiponectin was also significantly lower in tumor-bearing mice and this level was not decreased further by RT. Three fractions of RT appeared to decrease adiponectin levels in both normal and tumor-associated adipose tissue. However, the effect only reached statistical significance in the latter instance. The leptin/adiponectin ratio was increased in tumor-bearing mice, but RT had no effect on this ratio in either tumor-bearing or control mice [
118].
Tumor-bearing mice showed substantially decreased plasma levels of amylin, which is co-secreted with insulin by pancreatic β-cells [
118]. However, these levels were unaffected by RT in control and tumor-bearing mice. The plasma concentrations of hormones, which maintain glucose homeostasis and regulation of body weight, including insulin, glucagon, glucagon-like peptide, ghrelin, and pancreatic polypeptide, were not significantly lower in tumor-bearing versus control mice and these were not altered significantly by RT in either case.
Repeated activation of the ATX-LPA-inflammatory cycle should decrease the efficacy of RT by stimulating a wound healing response [
11,
123,
124]. First, RT-induced increases in expressions of ATX and activation of LPA2 receptors could decrease cancer cell death by depletion of the pro-apoptotic protein, Siva-1 [
11]. However, RT-induced apoptosis is less important in solid tumors compared to the effects on intestinal epithelial cells. The major therapeutic effect of RT for breast tumors is in causing some form of cytostasis (senescence or polyploid giant-cell formation) [
125]. In agreement with this, administration of the ATX inhibitor, GLPG1690, at the time of irradiation with five fractions of 7.5 Gy to breast tumors in mice decreased proliferation of breast cancer cells in the remaining tumor following the irradiation [
126]. This work supports the hypothesis that blocking the RT-induced activation of the ATX-LPA-inflammatory cycle can improve the efficacy of RT in eliminating residual cancer cells.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21165938