Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemical Research Methods
Calcium controls numerous biological processes by interacting with different classes of calcium binding proteins (CaBP’s), with different affinities, metal selectivities, kinetics, and calcium dependent conformational changes.
- rational design
- CaBP
- protein
- protein engineering
- synthesis
- fusion
1. Introduction
Ca2+ is the most ubiquitous signaling molecule in the human body, regulating numerous biological functions including heartbeat, muscle contraction, neural function, cell development, and cell proliferation. Calcium dynamics result from fluxes in Ca2+ concentrations that vary in amplitude and duration, between intracellular compartments and between the intracellular and extracellular space and intracellular organelles [1,2]. Temporal and spatial changes of Ca2+ concentrations in different cellular compartments affect the regulation of cellular signaling. Through signal stimulation or alteration in the membrane potential, the cytosolic Ca2+ concentration increases from 10−7 to 10−5 M, and vice-versa. These temporal and spatial changes of calcium concentrations and dynamics are orchestrated by various calcium-binding proteins (Figure 1).
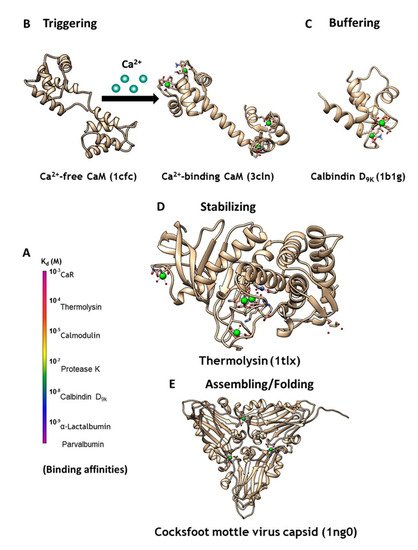
Figure 1. (A) CaBPs exhibit a broad range of binding affinities, which can vary by function. (B) Intracellular signaling protein calmodulin (CaM) binds up to four calcium ions as a secondary messenger. (C) Calbindin D9K acts as an intracellular calcium buffering protein. (D) Thermolysin is a zinc-activated metalloproteinase that is stabilized through binding of four calcium ions. (E) Calcium may also be required as a cofactor for assembling and folding of virus capsid, as seen in the Cocksfoot mottle virus.
Calcium modulating/triggering proteins such as calmodulin (CaM) are capable of transducing the intracellular Ca2+ signal changes into a number of diverse cellular events, including apoptosis, cell differentiation and cell proliferation. Through its cooperative binding to Ca2+ via multiple coupled calcium binding sites and calcium-dependent conformational changes, calcium trigger proteins can respond to narrow changes of calcium concentration. Of the myriad Ca2+-binding proteins that modulate cell signaling, CaM (Figure 1B) has the most versatile ability to respond to different cellular calcium concentrations through its two pairs of coordinated EF-Hand Ca2+-binding sites. Additionally, it activates or inhibits more than 300 functional enzymes, cellular receptors, and ion channels including ryanodine receptor and IP3 receptor at the ER, and gap junction proteins at the plasma membrane, through different binding modes [3,4]. Calcium buffering proteins such as calbindinD9K and parvalbumin act as buffer proteins (Figure 1C) to tightly control intracellular calcium concentrations gradients and kinetics. ER calcium buffer proteins such as calsequestrin with multiple calcium binding sites also controls ER-mediated calcium dynamics. Extracellular calcium concentration is operating at much high calcium concentration (1.1–2.2 mM). G-protein coupled receptors such as calcium sensing receptor and metabotropic glutamate receptors as well as proteins are able to bind to calcium and conduct/trigger subsequent signaling cascade [5,6,7,8]. Calcium binding also plays important structural roles for protein stabilization (Figure 1D), virus assembly and folding (Figure 1E), and to inhibit degradation [9]. However, the mechanisms through which different classes of proteins are able to bind calcium with different affinities (Figure 1A), and the resulting calcium dependent conformational changes, remain poorly understood. Efforts to design CaBP’s were hindered by the diversified coordination properties and binding geometries of CaBP’s, coupled with conformational changes of the proteins due to electrostatic interactions. In addition, there is an emerging need to develop calcium sensors capable of monitoring calcium dynamics and activity in different cellular environments to better understand the role of calcium signaling at cellular and molecular levels.
2. Classification and Prediction of Calcium Binding Sites in Proteins
To capture common features of calcium binding proteins and with a goal of visualizing the role of Ca2+ in biological systems (Calciomics), we performed a statistical analysis of various types of known Ca2+ binding proteins including EF-hand and non EF-hand proteins, and compared these results with properties of small molecules [16,17,18]. Results of these studies demonstrated that the structures and coordination geometries surrounding Ca2+ binding sites are more diverse than previously believed, and that Ca2+ binding sites in proteins are better reclassified as holospheric and hemispheric, instead of the classical polygon coordination reported in small molecule chelators. Ca2+ binding preferences of different amino acids were defined based on their observed frequency within the Ca2+ binding sites of proteins. Subsequently, several graph theory-based computational algorithms were developed that utilized our statistical data to predict Ca2+-binding sites with high accuracy and sensitivity in proteins using oxygen and carbon clusters [19,20,21,22]. Similarly, our lab has developed a search engine for predicting various classes of calcium binding proteins based on sequential pattern recognition [18,23]. These developed computer methods have been applied to predict calcium binding sites in bacteria and virus systems [9,24], as well as membrane receptors and transporters [8,25,26].
3. Development of Grafting and Design Approaches to Discover Key Factors that Contribute to Ca2+ Binding Affinities and Selectivities, and Ca2+-Induced Conformational Change
To overcome the complexities encountered in cooperative, multi-site binding associated with natural calcium binding proteins, two novel approaches (designing and grafting) have been developed to create single Ca2+-binding site sensor proteins. These structure present opportunities to dissect the key structural factors responsible for Ca2+-binding affinity, conformational change, and cooperativity (Figure 2).
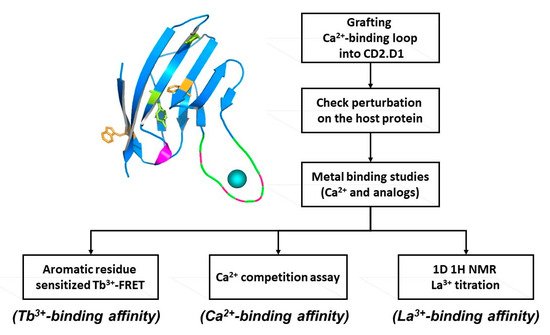
Figure 2. Determining intrinsic metal binding properties stepwise, through grafting of EF-loop (top left) into a scaffold protein (top right).
A single continuous calcium binding site like the EF-loop of CaM can be engineered into the loop region of a scaffold protein, like domain 1 of cell adhesion molecule CD2, which can tolerate mutations and has Trp residues. A flexible linker such as GGG is used to flank the engineered calcium binding site, which enables the Ca2+-binding loop to retain its native conformation for calcium binding. Intrinsic calcium binding affinity of the continuous Ca2+-binding site can be determined by bacterial expression and purification of the grafted protein. Tb3+, which has coordination properties similar to Ca2+, can be used to determine Tb3+ binding affinity via Tb-sensitized energy transfer. Calcium binding affinity is then determined by competition with the FRET enhancement. This method is very sensitive as the three positive charges on the Tb3+ ion exhibit higher metal binding affinity than Ca2+, and the use of Tb3+ has additional advantages over Ca2+ which often causes interference due to its ubiquitous background presence in solutions. Using this grafting approach we reported the first estimation of the intrinsic Ca2+ affinities of the four EF-hand loops of CaM, and a better estimation of the cooperativity of coupled EF-hand proteins [27,28,29,30]. This developed grafting approach has been successfully applied to obtain site-specific calcium binding affinities of our predicted single EF-hand motif in the non-structural protein of rubella virus [24]. Later, we further extended this grafting approach to probe site specific calcium binding affinity of non-EF-hand continuous calcium binding sites in G-protein coupled receptors (GPCR) including calcium sensing receptor [25,26] and mGluR1 [8]. Taking advantage of its relaxation properties, the lanthanide binding capability of EF-hand peptides have also been used to determine NMR (Nuclear Magnetic Resonance) structures [31].
Using identified common calcium coordination properties in proteins based on the statistical analysis and developed prediction algorithms, we successfully designed several de novo calcium-binding sites into the non-calcium binding protein CD2 (Figure 3) [32]. The NMR solution of the structure (1T6W) revealed that Ca.CD2 would bind Ca2+ in the designed site, which validated this general strategy for de novo design of Ca2+-binding proteins [33,34]. We have shown that Ca2+ and Ln3+-binding affinities, and protein stability, can be systematically tuned by altering metal coordination shells and thus the protein environment [35,36,37]. Using a single designed calcium binding site in the scaffold protein did not produce large conformational changes, thereby demonstrating that calcium binding affinity can be gradually increased by increasing the number of charged residues in the coordination shell from three to five. Additionally, to understand calcium dependent conformational changes, we also successfully designed a Ca2+-dependent CD2.trigger whose conformation could be reversibly switched upon Ca2+-binding without using coupled EF-hand motifs [38].
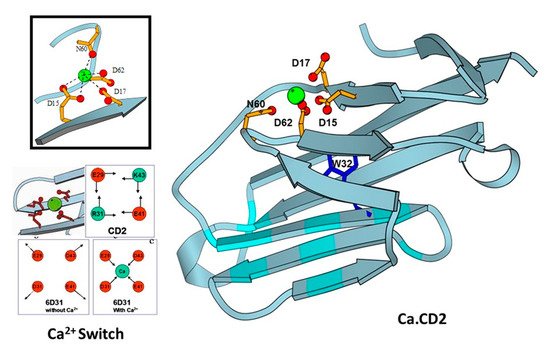
Figure 3. Design of CaBPs using cell adhesion molecule CD2 domain 1 with different arrangement of charged ligand residues. Determined NMR structure for Ca.CD2 (1T6W). Affinties were charge-dependent, with the highest affinity corresponding to the highest negative charge (−5 > −4 > −3 > −2).
4. Development of Genetically Encoded Calcium Indicators (GECIs) Using Protein Engineering
Building on the earlier work using aequorin to sense calcium, fluorescent proteins with intrinsic chromophore (FPs) of different colors have been developed for a variety of purposes, including uses as sensors, indicators of activity, and for enhanced microscopy visualization in cells and in tissues [39,40,41,42]. Genetically encoded calcium indicators (GECIs) have been widely used to elucidate various physiological mechanisms regulated by calcium signaling. Advanced GECIs provide quantitative analysis of Ca2+ fluctuations in different subcellular compartments which is essential to defining the mechanisms of Ca2+-dependent signaling under physiological and pathological conditions. Here we review the approaches for development of current GECIs and provide potential strategies for future development.
The endogenous Ca2+-trigger protein calmodulin (CaM), or troponin C (TnC), is commonly used as the calcium-binding domain to sense calcium changes for the design of most GECIs. Following cooperative binding to calcium, CaM or TnC exhibit additional conformational changes upon binding to its target proteins or peptides such as M13 peptide sequence from myosin light chain kinase (MLCK). This conformational change was first coupled with distance changes of two fluorescent proteins that have overlapping fluorescence excitation and emission wavelengths, producing a Forster resonance energy transfer (FRET) change. The development of calcium sensors was pioneered by Persichini [43] and Roger Tsien [1], whose subsequent work on engineering changes to GFP [44] led to a wide range of fluorescent protein applications. Improvements were then made to reduce interferences with intracellular CaM or CaM targeting proteins by modifying the CaM/peptide binding surface [45], or by modifying TnC to have narrowed specificity for its target protein TnI [46]. More recently, Barykina et al. reported development of a novel green Ca2+ indicator, FGCaMP, comprised of fungal CaM fused with EGFP, and a fragment of CaM-dependent kinase [47]. This novel green Ca2+ indicator was utilized to visual calcium transients in mammalian cells and observe calcium dynamics in neuronal cells with significantly improve mobility compared with other GECI’s.
5. Development of Calcium Sensors Using Rational Design
To overcome limitations associated with slow kinetics and non-linearity to CaM, Yang et al. pioneered another novel way to create calcium sensors by designing a single calcium binding site into a fluorescent protein without using endogenous Ca2+ binding proteins [76,77,78,79,80]. They first grafted a single EF-hand calcium binding motif to EGFP to create a calcium sensor Ca-G1-ER. The subsequent addition of calcium resulted in changes to both the absorbance and fluorescence emission spectra [80]. Detailed kinetic studies revealed that an intermediate state limits kinetics. To overcome rate limited steps by the formation of intermediate complex, CatchER, a calcium sensor for detecting high calcium concentrations, was generated by mutating five solvent-accessible residues on the surface of EGFP but in a location opposite to the chromophore, into negatively-charged residues, thereby forming a new Ca2+ binding site (Figure 4). Binding of Ca2+ to this site then triggered local conformational change to alter the hydrogen bonds surrounding the chromophore, resulting in changes to the fluorescent intensity. CatchER produced two absorption peaks at 395 nm and 488 nm. The Ca2+ binding process resulted in increased intensity of the 488 nm peak, with concurrent loss of signal intensity of the 395 nm peak. Additionally, excitation at either 395 nm or 488 nm resulted in higher peak intensity at 510 nm in the Ca2+ loaded form compared to the Ca2+ free form. Wild type GFP exhibits peaks at both 395 nm and 488 nm, while EGFP produces a single peak at 488 nm. The high-resolution X-ray crystal structure of CatchER suggested that the hydrogen network of the apo-form chromophore was similar to wild type GFP, whereas the Holo form appeared to mimic EGFP. These data suggested that the slight rotation of the Glu222 sidechain that occurred during transition from the Apo- to the Holo- form of CatchER played an important role in altering the chromophore hydrogen bonds.
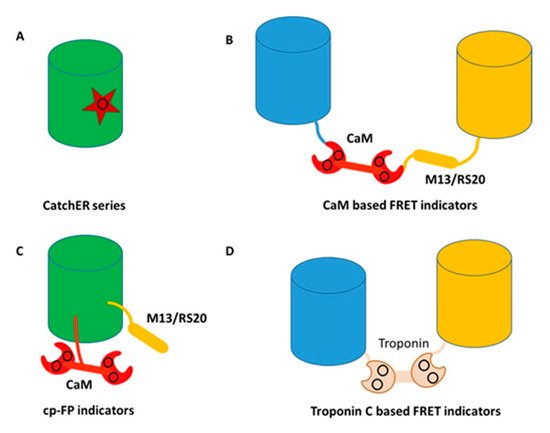
Figure 4. Classes of genetically encoded calcium indicators (GECIs) (A) rational design of a single calcium binding site, CatchER series. Star represents the binding site consisting of negatively charged residues; (B) CaM based FRET indicators. Blue and yellow cylinders represent blue and yellow fluorescent proteins, respectively. The black circle shows the calcium binding sites; (C) CaM based single wavelength indicators. The black circle shows the calcium binding sites; (D) Troponin C based FRET indicators. Blue and yellow cylinders represent blue and yellow fluorescent protein, respectively. The black circle shows the calcium binding sites.
This entry is adapted from the peer-reviewed paper 10.3390/molecules25092148
This entry is offline, you can click here to edit this entry!