The emergence of a nucleosome-based chromatin structure accompanied the evolutionary transition from prokaryotes to eukaryotes. In this scenario, histones became the heart of the complex and precisely timed coordination between chromatin architecture and functions during adaptive responses to environmental influence by means of epigenetic mechanisms. Notably, such an epigenetic machinery involves an overwhelming number of post-translational modifications at multiple residues of core and linker histones.
- histone post-translational modifications
- chromatin
- nucleosome
- epigenetics
1. Introduction
Chromatin is a polymeric nucleoprotein complex composed primarily of DNA closely associating with positively charged histone proteins [1]. The basic structural repeating unit is the nucleosome core particle, consisting of 147 base pairs of DNA wrapped nearly twice in a left-handed toroidal supercoil around a core histone octamer [2]. The latter exhibits a tripartite organization, which comprises a tetramer containing two copies of H3 and H4, coupled to two H2A-H2B dimers [3]. Each core histone contains a globular histone fold domain, which mediates heterodimeric interactions, and two amino- and carboxy-terminal unstructured extensions protruding from the surface of the nucleosome [4]. These flexible domains, commonly known as the histone tails, provide additional stabilization effects through their contacts with the nucleosomal DNA backbone and acidic patch of adjacent nucleosomes [5]. As an additional hierarchical level of chromatin compaction, core particles are further stabilized by the binding of histone H1 at the two linker DNA arms entering and exiting on the surface of each octamer [6][7]. Molecular processes establishing such a histone stoichiometry determines the basic chromatin fiber structure [8][9].
Key mechanisms contributing to the dynamic regulation of the degree of chromatin compaction to allow the harmonious utilization of the genetic information include covalent post-translational modifications (PTMs) of histones [10][11]. In this regard, histone tails represent preferential, but not exclusive, targets of a myriad of PTMs that correlate with changes in gene activity. In fact, the recent improvement of the sensitivity of mass spectrometry-based methods allowed the discovery of more than two hundred histone modification sites so far ( Figure 1 ), although no functional significance has been assigned to the majority of them [12][13]. These modifications are imposed, and can be reversed as well, by an assortment of histone-modifying enzymes, so that they represent a major mechanism of cellular adaptation to the varying environmental conditions [[1][14][15][16]. Broadly speaking, the presence of histone PTMs alters the stereochemical environment of the nucleosomes and their binding affinity for regulatory complexes that can be recruited or turned away from chromatin, thereby eliciting functional outcomes.
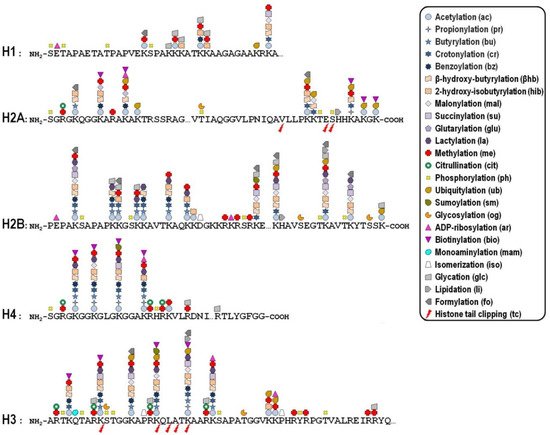
Whether a given histone PTM correlates with gene expression in a positive or negative fashion most likely depends on its genomic location. To make matters more complicated, it is now becoming clear that the combinatorial nature of histone PTMs implies both the coalescence of multiple modifications on the same nucleosome and the possibility of functional promiscuity induced by synergistic or antagonistic cross-talk among them. The simplest example of such a cross-talk is the direct competition, on the same histone residue, among alternative PTMs associated with functionally opposite transcriptional readouts. In many other cases, one modification promotes/inhibits the generation/removal of another modification “in cis”, viz within a distinct site in the same histone, or “in trans”, between distinct histones either within the same nucleosome or across nucleosomes, as well as between histones and DNA methylation [17]. In addition, different histone PTMs may cooperate in order to recruit specific complexes efficiently [18].
2. Acetylation
In the early ‘60s, Phillips pioneered the field of histone PTMs originally reporting the N-acetylation of lysine residues in histones extracted from calf thymus [19]. Curiously, many authors attributed the achievement of this discovery to Allfrey and collaborators, although they clearly gave credit to Phillips in their first paper on this topic [20]. Undoubtedly, acetylation became the most studied histone PTM, so that nowadays we know that selected lysines are acetylated during specific biological processes, and that the overall histone acetylation level is dynamically modulated by the opposing action of two families of enzymes, namely the histone acetyltransferases (HATs) and the histone deacetylases (HDACs). This nomenclature, albeit currently used, is actually inappropriate because these enzymes can respectively transfer or remove a wide repertory of acyl species, besides the acetyl group, to both histone and non-histone target proteins (see below). In the case of acetylation, the HATs require acetyl-CoA as a cofactor to catalyse the transfer of an acetyl group to the ε-amino group of lysine side chains [21]. Based on their subcellular localization, HATs have been traditionally divided in two distinct types: type-A HATs are directly involved in histone acetylation in the context of nucleosomes, while type-B HATs acetylate free newly synthesized histones in the cytoplasm to facilitate their incorporation into the newly replicated chromatin [22][23]. HDAC enzymes reverse the activity of HATs, restoring the original unacetylated state of histone lysines. There are four classes of HDACs, whose members typically associate into multiprotein complexes showing rather low substrate specificity [24]. Collectively, both the mentioned enzyme families modify numerous, but not all, histone lysines in vivo [25]. Importantly, the HAT activity, differential substrate specificity and recruitment are modulated through multisubunit complex formation [26]. For example, yeast Gcn5 alone does acetylate free histones in vitro, but only when it is combined with the Ada2 and Ada3 subunits within the ADA and SAGA complexes, it can preferentially modify H2B enclosed into nucleosomes [27].
In principle, the observation that acetylation of lysine residues neutralizes their positive charge would suggest that HATs may reduce chromatin compaction by weakening the stabilizing influence of histone-DNA and inter-nucleosomal electrostatic interactions [28]. Accordingly, acetylation of H4K16 has been demonstrated to directly impact on chromatin structure by charge neutralization and loss of H4 tail flexibility, collectively reducing interaction with the neighbouring nucleosome [29][30][31]. In support of this evidence, ChIP-seq studies on a genome-wide scale revealed that site-specific acetylation is frequently associated with enhancers and promoters of actively transcribed genes [32][33][34][35]. However, there is some variation in the exact distribution of individual acetylation sites along the gene structure. For instance, H3K9ac peaks at the transcription start site, while H4K16ac shows a broader localization across gene bodies [35]. In sharp contrast, structural studies highlighted that lysine acetylation itself generally does not provoke major reorganization of DNA or core histones at the nucleosome level [36], leading to reasonably suppose that histone acetylation could generate recognition sites for effector proteins involved in the local regulation of chromatin organization and transcriptional activity.
3. Methylation
Unlike acetylation, histone methylation does not alter the net charge of modified residues and involves additional layers of complexity, because the side chains of two distinct amino acids can be modified at a different extent. More specifically, the ε-amino group of lysines can be mono-, di- or tri-methylated, while the ω-guanidine group of arginines can be mono- or di-methylated either symmetrically or asymmetrically [37][38].
Histone methyltransferases (HMTs) and demethylases ( HDMs) enzymes carry out fine-tuning of the methylation state. Based on the specificity towards lysine or arginine, two major classes of HMTs can be distinguished, each of them containing a distinctive catalytic domain that transfers a methyl group from the S-adenosylmethionine donor. In contrast to other histone modifying enzymes, members of both classes are devoted to methylation of highly specific residues to an appropriate degree. For example, PRMT7 catalyses the formation of only mono-methyl-arginine, while PRMT5 electively determines symmetrical di-methylation of arginine [39][40]. Similarly, DIM5 univocally targets H3K9 for tri-methylation, while SET7/9 can only mono-methylate H3K4 in Neurospora crassa [41][42]. Notably, unlike acetylation, the different methylated histone sites may associate with diametrically opposed functional outcomes [43]. For instance, H3K4me3 and H3K36me3 have been primarily linked to a transcriptionally permissive chromatin environment [44], H3K4me1 is considered a predictive hallmark of active enhancers [45], while H3K27me3 and H4K20me3 have been generally associated with heterochromatin formation and gene silencing [46][47]. Interestingly, H3K9 provides an emblematic example of the dualistic association of methylation with repressed and active states of chromatin. Indeed, several studies concordantly showed that H3K9me3 represents a docking site for the α isoform of the heterochromatin protein 1 (HP1α), which assists the progress of chromatin condensation in various organisms [48][49]. Other studies came to apparently conflicting conclusions, demonstrating that H3K9me3 and the HP1γ isoform co-localize in mammalian euchromatic regions, where they are involved in transcriptional elongation and are rapidly lost upon cessation of transcription [50].
Analogously to other epigenetic mechanisms, the histone methylation status varies widely during adaptive responses to environmental influence. For example, H3K14me3 is not normally found in human chromatin but it is distinctively generated following infection of lung epithelial cells with the intracellular pathogen Legionella pneumophila [51].
As with the HMTs, HDM enzymes display a high grade of target specificity, being sensitive to the degree of methylation. For example, JMJD2A can reverse H3K9me3 to H3K9me2 but not to H3K9me1 [52]. Removal of methyl groups from mono-methylated and asymmetrically di-methylated arginines is most probably carried out by JMJD6 [53]. To date, this is the one and only enzyme possessing arginine demethylase activity directed toward histones, although this function has been questioned by contradictory evidence [54][55].
This entry is adapted from the peer-reviewed paper 10.3390/genes12101596
References
- Kornberg, R.D. Chromatin structure: A repeating unit of histones and DNA. Science 1974, 184, 868–871.
- Kornberg, R.D.; Lorch, Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 1999, 98, 285–294.
- Arents, G.; Burlingame, R.W.; Wang, B.C.; Love, W.E.; Moudrianakis, E.N. The nucleosomal core histone octamer at 3.1 Å resolution: A tripartite protein assembly and a left-handed superhelix. Proc. Natl. Acad. Sci. USA 1991, 88, 10148–10152.
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260.
- Ausio, J.; Dong, F.; van Holde, K.E. Use of selectively trypsinized nucleosome core particles to analyze the role of the histone “tails” in the stabilization of the nucleosome. J. Mol. Biol. 1989, 206, 451–463.
- Bednar, J.; Horowitz, R.A.; Grigoryev, S.A.; Carruthers, L.M.; Hansen, J.C.; Koster, A.J.; Woodcock, C.L. Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc. Natl. Acad. Sci. USA 1998, 95, 14173–14178.
- Bednar, J.; Garcia-Saez, I.; Boopathi, R.; Cutter, A.R.; Papai, G.; Reymer, A.; Syed, S.H.; Lone, I.N.; Tonchev, O.; Crucifix, C.; et al. Structure and Dynamics of a 197 bp Nucleosome in Complex with Linker Histone H1. Mol. Cell 2017, 66, 384–397.
- Cavalieri, V.; Melfi, R.; Spinelli, G. Promoter activity of the sea urchin (Paracentrotus lividus) nucleosomal H3 and H2A and linker H1 -histone genes is modulated by enhancer and chromatin insulator. Nucleic Acids Res. 2009, 37, 7407–7415.
- Cavalieri, V.; Melfi, R.; Spinelli, G. The Compass-like locus, exclusive to the Ambulacrarians, encodes a 874 chromatin insulator binding protein in the Sea Urchin embryo. PLoS Genet. 2013, 9, e1003847.
- Cavalieri, V.; Spinelli, G. Histone-mediated transgenerational epigenetics. In Transgenerational Epigenetics, 2nd ed.; Tollefsbol, T.O., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 157–183.
- Cavalieri, V. Histones, Their Variants and Post-translational Modifications in Zebrafish Development. Front. Cell Dev. Biol. 2020, 8, 456.
- Su, X.; Ren, C.; Freitas, M.A. Mass spectrometry-based strategies for characterization of histones and their posttranslational modifications. Expert Rev. Proteom. 2007, 4, 211–225.
- Zhao, Y.; Garcia, B.A. Comprehensive Catalog of Currently Documented Histone Modifications. Cold Spring Harb. Perspect. Biol. 2015, 7, a025064.
- Smith, E.; Shilatifard, A. The chromatin signaling pathway: Diverse mechanisms of recruitment of histone-modifying enzymes and varied biological outcomes. Mol. Cell 2010, 40, 689e701.
- Cavalieri, V.; Spinelli, G. Environmental epigenetics in zebrafish. Epigenetics Chromatin 2017, 10, 46.
- Cavalieri, V. Model organisms and their application in environmental epigenetics. In Environmental Epigenetics in Toxicology and Public Health; Fry, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 67–87.
- Fischle, W.; Wang, Y.; Allis, C.D. Binary switches and modification cassettes in histone biology and beyond. Nature 2003, 425, 475–479.
- Oey, N.E.; Leung, H.W.; Ezhilarasan, R.; Zhou, L.; Beuerman, R.W.; VanDongen, H.M.; VanDongen, A.M. A Neuronal Activity-Dependent Dual Function Chromatin-Modifying Complex Regulates Arc Expression. eNeuro 2015, 2, ENEURO.0020-14.2015.
- Phillips, D.M.P. The presence of acetyl groups in histones. Biochem. J. 1963, 87, 258–263.
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794.
- Berndsen, C.B.; Denu, J.M. Catalysis and substrate selection by histone/protein lysine acetyltransferases. Curr. Opin. Struct. Biol. 2008, 18, 682–689.
- Richman, R.; Chicoine, L.G.; Collini, M.P.; Cook, R.G.; Allis, C.D. Micronuclei and the cytoplasm of growing Tetrahymena contain a histone acetylase activity which is highly specific for free histone H4. J. Cell Biol. 1988, 106, 1017–1026.
- Parthun, M.R.; Widom, J.; Gottschling, D.E. The major cytoplasmic histone acetyltransferase in yeast: Links to chromatin replication and histone metabolism. Cell 1996, 87, 85–94.
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713.
- Thorne, A.W.; Kmiciek, D.; Mitchelson, K.; Sautiere, P.; Crane-Robinson, C. Patterns of histone acetylation. Eur. J. Biochem. 1990, 193, 701–713.
- Kimura, A.; Horikoshi, M. How do histone acetyltransferases select lysine residues in core histones? FEBS Lett. 1998, 431, 131–133.
- Grant, P.A.; Duggan, L.; Côté, J.; Roberts, S.M.; Brownell, J.E.; Candau, R.; Ohba, R.; Owen-Hughes, T.; Allis, C.D.; Winston, F. Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: Characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes Dev. 1997, 11, 1640–1650.
- Workman, J.L.; Kingston, R.E. Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu. Rev. Biochem. 1998, 67, 545–579.
- Shogren-Knaak, M. Histone H4–K16 acetylation controls chromatin structure and protein interactions. Science 2006, 311, 844–847.
- Allahverdi, A.; Yang, R.; Korolev, N.; Fan, Y.; Davey, C.A.; Liu, C.F.; Nordenskiöld, L. The effects of histone H4 tail acetylations on cation-induced chromatin folding and self-association. Nucleic Acids Res. 2011, 39, 1680–1691.
- Collepardo-Guevara, R.; Portella, G.; Vendruscolo, M.; Frenkel, D.; Schlick, T.; Orozco, M. Chromatin unfolding by epigenetic modifications explained by dramatic impairment of internucleosome interactions: A multiscale computational study. J. Am. Chem. Soc. 2015, 137, 10205–10215.
- Schubeler, D.; MacAlpine, D.M.; Scalzo, D.; Wirbelauer, C.; Kooperberg, C.; van Leeuwen, F.; Gottschling, D.E.; O’Neill, L.P.; Turner, B.M.; Delrow, J.; et al. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev. 2004, 18, 1263–1271.
- Cavalieri, V.; Spinelli, G. Ectopic hbox12 Expression Evoked by Histone Deacetylase Inhibition Disrupts Axial Specification of the Sea Urchin Embryo. PLoS ONE 2015, 10, e0143860.
- Di Caro, V.; Cavalieri, V.; Melfi, R.; Spinelli, G. Constitutive promoter occupancy by the MBF-1 activator and chromatin modification of the developmental regulated sea urchin alpha-H2A histone gene. J. Mol. Biol. 2007, 365, 1285–1297.
- Wang, Z.B.; Zang, C.Z.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903.
- Brower-Toland, B.; Wacker, D.A.; Fulbright, R.M.; Lis, J.T.; Kraus, W.L.; Wang, M.D. Specific contributions of histone tails and their acetylation to the mechanical stability of nucleosomes. J. Mol. Biol. 2005, 346, 135–146.
- Ng, S.S.; Yue, W.W.; Oppermann, U.; Klose, R.J. Dynamic protein methylation in chromatin biology. Cell Mol. Life Sci. 2009, 66, 407–422.
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 2009, 33, 1–13.
- Zurita-Lopez, C.I.; Sandberg, T.; Kelly, R.; Clarke, S.G. Human protein arginine methyltransferase 7 (PRMT7) is a type III enzyme forming ω-NG-monomethylated arginine residues. J. Biol. Chem. 2012, 287, 7859–7870.
- Wang, M.; Xu, R.M.; Thompson, P.R. Substrate specificity, processivity, and kinetic mechanism of protein arginine methyltransferase 5. Biochemistry 2013, 52, 5430–5440.
- Tamaru, H.; Zhang, X.; McMillen, D.; Singh, P.B.; Nakayama, J.; Grewal, S.I.; Allis, C.D.; Cheng, X.; Selker, E.U. Trimethylated lysine 9 of histone H3 is a mark for DNA methylation in Neurospora crassa. Nat. Genet. 2003, 34, 75–79.
- Xiao, B.; Jing, C.; Wilson, J.R.; Walker, P.A.; Vasisht, N.; Kelly, G.; Howell, S.; Taylor, I.A.; Blackburn, G.M.; Gamblin, S.J. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature 2003, 421, 652–656.
- Zhang, Y.; Reinberg, D. Transcription regulation by histone methylation: Interplay between different covalent modifications of the core histone tails. Genes Dev. 2001, 15, 2343–2360.
- Schneider, R.; Bannister, A.J.; Myers, F.A.; Thorne, A.W.; Crane-Robinson, C.; Kouzarides, T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat. Cell Biol. 2004, 6, 73–77.
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318.
- Noma, K.; Allis, C.D.; Grewal, S.I. Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science 2001, 293, 1150–1155.
- Schotta, G.; Lachner, M.; Sarma, K.; Ebert, A.; Sengupta, R.; Reuter, G.; Reinberg, D.; Jenuwein, T. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004, 18, 1251–1262.
- Lachner, M.; O’Carroll, D.; Rea, S.; Mechtler, K.; Jenuwein, T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001, 410, 116–120.
- Maison, C.; Almouzni, G. HP1 and the dynamics of heterochromatin maintenance. Nat. Rev. Mol. Cell Biol. 2004, 5, 296–304.
- Vakoc, C.R.; Mandat, S.A.; Olenchock, B.A.; Blobel, G.A. Histone H3 lysine 9 methylation and HP1gamma are associated with transcription elongation through mammalian chromatin. Mol. Cell 2005, 19, 381–391.
- Rolando, M.; Sanulli, S.; Rusniok, C.; Gomez-Valero, L.; Bertholet, C.; Sahr, T.; Margueron, R.; Buchrieser, C. Legionella pneumophila effector RomA uniquely modifies host chromatin to repress gene expression and promote intracellular bacterial replication. Cell Host Microbe 2013, 13, 395–405.
- Whetstine, J.R.; Nottke, A.; Lan, F.; Huarte, M.; Smolikov, S.; Chen, Z.; Spooner, E.; Li, E.; Zhang, G.; Colaiacovo, M.; et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 2006, 125, 467–481.
- Chang, B.; Chen, Y.; Zhao, Y.; Bruick, R.K. JMJD6 is a histone arginine demethylase. Science 2007, 318, 444–447.
- Webby, C.J.; Wolf, A.; Gromak, N.; Dreger, M.; Kramer, H.; Kessler, B.; Nielsen, M.L.; Schmitz, C.; Butler, D.S.; Yates, J.R., 3rd; et al. Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science 2009, 325, 90–93.
- Mantri, M.; Krojer, T.; Bagg, E.A.; Webby, C.A.; Butler, D.S.; Kochan, G.; Kavanagh, K.L.; Oppermann, U.; McDonough, M.A.; Schofield, C.J. Crystal structure of the 2-oxoglutarate- and Fe(II)-dependent lysyl hydroxylase JMJD6. J. Mol. Biol. 2010, 401, 211–222.