1. Introduction
We live surrounded by environmental hazards including toxic compounds in air, water, soil, and food. Pollution is ubiquitous and mostly unavoidable. Neurotoxic organic and inorganic compounds come from fossil fuel combustion, engineered nanoparticles, nanoplastics, and compounds resulting from disasters such as forest wildfires. We are constantly exposed to these environmental hazards, regardless of age, sex, or socioeconomic status. Some people are more vulnerable than others and the brain in development is a target. The complexities of our modern-day, fossil fuel-based society are a factor when we talk about central nervous system (CNS) effects and the role of specific pollutants.
Minding only criteria pollutants [1] does not give us the full picture of exposure risk because these six compounds are regulated primarily for meeting regional air quality standards, rather than explicitly for reducing chronic or acute exposures and causality of adverse health outcomes. Other policies and standards exist for limiting pollutant-specific near-source exposures. Furthermore, criteria pollutants measured in ambient air (ozone (O 3), particulate matter (PM), sulfur dioxide (SO 2), nitrogen dioxide (NO 2), carbon monoxide (CO), and lead (Pb)) are operationally defined. Comparisons of pollution burdens across regions and countries must begin by ensuring the application of identical measurement methods and analytical techniques, otherwise we risk comparing apples to oranges. In addition, PM and O 3 are not singular entities; rather, they are lumped parameters representing a family of source-dependent constituents and precursors that can vary greatly in space and time and in physicochemical and toxicological characteristics. For these reasons, it is extremely difficult to make comparisons of PM 2.5 pollution and health effects in, for example, Delhi versus New York City or Provo, Utah versus Metropolitan Mexico City. Moreover, the determinants of adverse health outcomes are numerous: the strength and nature of emission sources, exposure times, and cumulative exposures over a lifetime, age, morbidities, and occupational history are some of the most obvious. The cumulative effect of these and other factors will determine specific health outcomes and responses vary significantly by population in terms of genetics, nutrition, exercise patterns, and cultural factors. These arguments should be an alarm for the reader. The sophistication of instruments, methodologies, and models to characterize air pollutants, exposures, or the results and interpretation of a specific PM 2.5 study, for example, may not equate to ability to understand and compare results, not even within the same country, let alone across the globe. Thus, local capacity to understand emission sources, implement PM control measures, and establish clinical, laboratory, and pathology links becomes paramount.
There is strong evidence of causality between PM 2.5 air pollution exposure and cardiovascular morbidity and mortality [2]. The 2021 WHO Global Air Quality Guidelines recommendation for annual PM 2.5 Air Quality Guideline Level is 5 µg/m 3, with four interim targets proposed as incremental steps in a progressive reduction of air pollution and intended for use in highly polluted areas [3]. General practitioners, emergency room doctors, internists, neurologists, psychiatrists, pediatricians, cardiologists, infectologists (think COVID), and others see firsthand the morbidity and mortality effects of air pollution and need readily available information about how to interpret results across locations.
2. Portals of Entry to the Brain and Key Neural Damage Mechanisms
The respiratory tract is a key portal of entry through inhalation as the air passes through the nasal cavity, directly to the olfactory nerve and olfactory bulb, the trigeminal, facial, and glossopharyngeal nerves, and to the lower respiratory tract and the alveoli [
14]. UFPs accumulated in children’s nasal epithelium, the olfactory bulb, and frontal cortex and extensive damage of the neurovascular unit (NVU) are documented in highly exposed Metropolitan Mexico City (MMC) young residents: capillary leakage, vascular amyloid, amyloid plaques, and hyperphosphorylated tau (P-tau) accumulation are present in the first two decades of life [
15]. The gastrointestinal (GI) system is an important portal through ingestion: we swallow a significant amount of the inhaled UFPs and ingest NPs added to food ingredients, supplements, food and drink containers, toothbrushes, and toothpaste. Titanium dioxide and silicon dioxide NPs are widely used as additives in the food industry and their long-time dietary intake produces gut microbiota disruption, intestinal, kidney, spleen, and liver injury, and neurotoxic effects through gut–brain axis disruption, with a major impact on children [
16]. Swallowed NPs/UFPs have easy access to the GI epithelium and submucosa and their damage allows direct access to the enteric nervous system [
17]. In a combined study of gastric, small bowel, and vagal nerves at the cervical level in children, young adults, and dogs with low versus high exposures to PM
2.5, NPs were abundant in erythrocytes, unmyelinated submucosal, perivascular, and intramuscular nerve fibers, ganglionic neurons, and vagal nerves and associated with organelle pathology, in highly exposed urbanites [
17]. Immunohistochemistry showed hallmarks of Parkinson’s and Alzheimer’s diseases including aggregated alpha-synuclein and P-tau in gastrointestinal tract and vagal nerves of young children and Mexico City young adults [
17]. The skin and mucosae are also critical portals of entry especially in certain occupational environments (fire—nanoparticle extinguishing agents), and in people using UV screen protectors [
12].
Once NPs reach the systemic circulation, they can travel freely or hitchhike on red or white blood cells and start their journey through the body [
14,
18]. The NVU [
19] is an early NP target and since the blood–brain barrier (BBB) breakdown contributes to blood barrier dysfunction, vascular leaks, and associated cognitive decline, the extensive NVU damage associated with NPs and documented in urban toddlers is likely an early step in the brain damage cascade [
14,
15]. Damage to the NVU complex functional and anatomical structure impacts endothelium, pericytes, astrocytes, microglia, capillaries, arterioles, basal lamina covered by pericytes, and neural cells including neurons, interneurons, and extracellular matrix [
19]. NVU cells and matrixes interact closely for maintaining homeostasis of neurovascular coupling, BBB integrity, and trans-endothelial fluid transport. Any disruption of the NVU will translate into failure of tight coupling between neural activity and cerebral blood flow (CBF) [
19]. Damage to red blood cells (RBCs) and brain endothelium associated with NPs [
14] is clearly contributing to vasopathological effects and compromising the interaction between RBCs, endothelial cells, platelets, and macrophages, and ultimately, as Petrini and coworkers [
20] discussed, altering thrombosis, hemostasis, and immune responses. NVU alterations predict brain dysfunction, neuroinflammatory responses, and neurodegeneration.
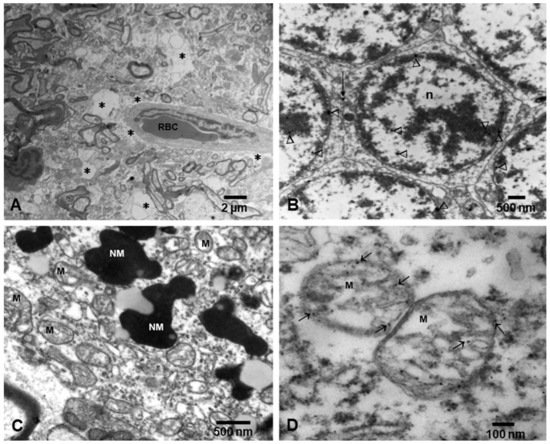
Dysfunctional mitochondria are an early feature of neurodegenerative diseases [
21] and early responses to excessive production of reactive oxygen species (ROS) result in the induction of mitophagy to remove damaged mitochondria. Mitochondrial damage associated with sustained oxidative stress, as in the exposure to air pollutants and NPs, overwhelms autophagic and mitophagic pathways, activating a vicious circle that leads to reduced capacity to remove damaged mitochondria, and/or an alteration in the regulation of mitophagy [
21]. Defective mitophagy leads to synaptic degeneration and mitochondrial fragmentation in association with Aβ and P-tau and cognitive dysfunction in Alzheimer’s disease (AD) [
21]. NPs are very effective in targeting the cell powerhouse in neural cells in air pollution-exposed children and young adults (
Figure 2).
Figure 2. Electron micrographs of neurovascular unit (NVU) and neural organelles in Metropolitan Mexico City children. (A) Three-year-old boy, substantia nigrae pars compacta showing a capillary with one luminal red blood cell (RBC) surrounded by an extensively vacuolated, fragmented neuropil (*). (B) Cerebellar granular neurons from same child as (A), showing nanoparticles (arrowheads) in intranuclear location and at the membrane interphase between neurons (short arrow). (C) Fifteen-year-old substantia nigrae pars compacta showing numerous mitochondria (M) with abnormal cristae and neuromelanin structures (NM) with nanoparticles. (D) In a closeup, the mitochondria exhibit numerous nanoparticles in the matrix, cristae, and along the double layer mitochondrial wall (arrows).
A major pathway involved in NPs’ detrimental brain effects is ROS production [
22]. The oxidative reactivity of NPs—over other physicochemical properties—causes cytotoxicity via induction of cellular oxidative stress, and a good example is combustion-associated Fe oxide NPs, both magnetite and maghemite, which once in a cell’s acidic lysosomal environment release free Fe ions [
22]. Inflammatory responses are followed by anti-inflammatory actions, in response to metal base NPs, including superparamagnetic Fe oxide NPs, and autophagy and impaired neovascularization are serious and unwanted neural responses [
22]. The size of NPs and chemical composition are critical for their blood circulation time, blood vessel damage, and neural toxicity [
22]. Gutiérrez et al. [
23] used 14 and 22 nm magnetic NPs with the same core but subjected to different surface modifications procedures; the results were NPs with different size aggregates and arrangements impacting their magnetic and heating properties. Gutiérrez and coworkers’ observations [
23] are critical for environmental magnetic NPs and brain effects: (1) colloidal stability depends on the balance of magnetic, dipolar, and van der Waals forces and repulsive interactions, mainly electrostatic and steric, and (2) when magnetic cores are assembled, the distance between cores affects magnetic behavior of the entire particle, i.e., dipole–dipole interactions result in an alteration of magnetic properties. In a live organism where NPs can be highly concentrated within endosomes, Gutiérrez et al. suggested magnetic interactions become very strong and exposure to alternating magnetic fields (AMFs) gives rise to heat produced by NPs and mainly determined by size, shape, crystal structure, saturation magnetization, and susceptibility [
23]. This information is very relevant to urban dwellers with significant concentrations of magnetic NPs in their brains [
14].
All NPs in biological fluids are surrounded by a corona and protein corona–NP binding affinity depends on shape, size, and surface characteristics of NPs and hydrodynamic, electrodynamic, and magnetic forces [
22,
23,
24]. High-affinity proteins forming coronas are stable, and protein adsorption on the surface of NPs will allow the protein to keep most of their native conformation, but as a result, the protein could be thermodynamically unstable. The protein adsorption depends on the available protein concentration and in environments with several proteins, the NP surface may bind several of them [
24]. It is critical to remember that the interaction of NPs with organelles occurs mostly through the NP–corona [
24], i.e., the same NP coated with different proteins will have easier access to a cell if the adsorbed protein unfolding helps in accessing cell surface receptors or inhibited if the protein structure is lost. As a result, the protein corona, the interaction of each individual protein, and the NP size influence larger surface concentration, cellular uptake, accumulation, degradation, and clearance of the NP. NPs can act as molecular chaperones to carry out protein folding, destabilization, and protein aggregates and as a result of elevation of protein concentrations and destabilization of protein, folded states promote amyloid aggregates [
24,
25].
The molecular chaperone function and the cellular efficiency of the degradation systems are key for the correct folding of the cellular protein and for preventing aggregation. Certain surfaces are very effective in promoting amyloid formation (i.e., those with lipid bilayers, collagen fibers, and polysaccharides). Hartl’s work [
25] emphasized that misfolded proteins expose hydrophobic amino acid residues and regions of unstructured polypeptide backbone, resulting in cytosolic protein aggregates rich in β-sheet structures, i.e., α-synuclein and tau. The common denominator of amyloid-prone proteins is their small size and the fact that are intrinsically disordered in non-aggregated states [
25].
We also need an intact endoplasmic reticulum (ER) for eliminating abnormal proteins, thus any ER alterations will result in a protein degradation failure. Highly exposed urban megacity residents have severe alterations in the ER, widespread degenerated mitochondria–ER contacts (MERCs), and quadruple protein misfolding in close association with NPs [
26].
In the scenario of PM
2.5 pollution and NPs, the current literature supports the following: (1) environmental NPs can directly alter brain proteins [
14,
15,
16,
17,
22,
23,
24]; (2) since NPs can travel anterograde, retrograde, and trans-synaptically from any portal of entry, altered proteins are capable of moving in all directions [
14,
15,
17]; (3) protein misfolding, aggregation, and fibrillation take place in neural cells and follow the NP path’s initial portal of entry (i.e., brainstem) [
17,
22,
23,
24]; and (4) neuroinflammation, damage to the neurovascular unit, oxidative stress, and magnetic effects all potentially contribute to neural damage [
22,
23,
24,
25,
26]. NPs can potentially elicit key mechanistic pathways as described in all major neurodegenerative diseases and produce the neuropathological features of several diseases at the same time, as we are observing in Mexico City children and young adults (
Figure 3) [
14,
15,
17,
26].
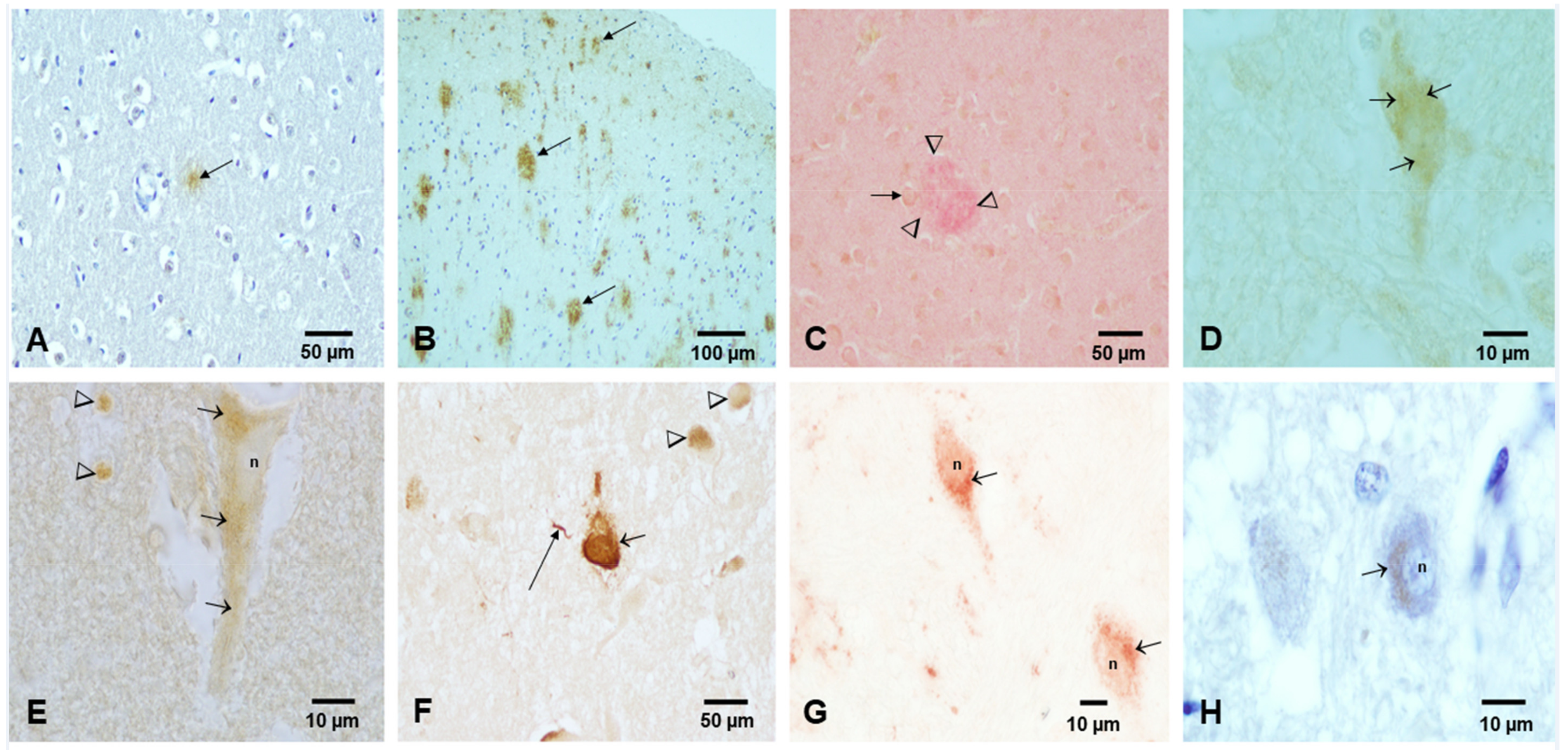
Figure 3. Light microscopy, immunohistochemstry (IHC) of aberrant neural proteins in MMC children and young adults. (A) Frontal cortex in an 11-month-old APOE 3/3 showing an Aβ diffuse plaque. IHC Aβ 17–24, 4G8, Covance, Emeryville, CA 1:1500, DAB brown product. (B) Temporal cortex, 11 y old child APOE 3/3 with multiple trans-cortical diffuse and mature Aβ plaques, 4G8 brown product. (C) Adult 36 y old, APOE 3/4, temporal cortex with mature amyloid plaques (arrowheads) and reactive astrocytes (short arrow). Double staining 4G8 red product, and glial acidic fibrillary protein (GAFP) brown product. (D) Two-year-old frontal cortex with granular cytoplasmic staining for hyperphosphorylated tau (P-tau) (short arrows). PHF-tau 8 phosphorylated at Ser 199–202-Thr 205, Innogenetics, Belgium, AT-8 1:1000, brown DAB product. (E) Eleven-year-old frontal cortex with pyramidal neuron granular cytoplasmic immunoreactivity for P-tau. (F) Forty-year-old male with numerous tangles supra and infratentorial, including substantia nigrae (short arrow), P-tau neurite (long arrow), and granular cytoplasmic immunoreactivity (arrowheads). AT-8 brown product. (G) Same 11 y old child as (E) with TPD-43 immunoreactivity in brainstem neurons, the nuclei (n) are negative, and the cytoplasm exhibits granular immunoreactivity (short arrows). TDP-43 red product. (H) Thirty-five-year old male, cochlear nuclei neurons positive for α-synuclein, phosphorylated at Ser-129, LB509, In Vitrogen, Carlsbad CA 1:1000. Red product counterstained with hematoxylin.
This entry is adapted from the peer-reviewed paper 10.3390/ijerph182111568