Flecainide is an IC antiarrhythmic drug approved in 1984 from Food and Drug Administration for the suppression of sustained ventricular tachycardia and later for acute cardioversion of atrial fibrillation (AF) and for sinus rhythm maintenance. It is categorized as a Vaughn-Williams Class IC agent based upon its properties of causes a strong degree of sodium channel blockage with slowing cardiac conduction and a minimal effect on ventricular repolarization. Currently, flecainide is mostly used for sinus rhythm maintenance in atrial fibrillation patients without structural cardiomyopathy although recent studies enrolling different patient population demonstrated a good effectiveness and safety profile.
- flecainide
- flecainide controlled-release
- atrial fibrillation
- CAST
- supraventricular arrhythmias
1. Introduction
Flecainide is a class IC antiarrhythmic drug (AAD). Flecainide was first synthesized in 1972 and approved in 1984 from the Food and Drug Administration (FDA) for the suppression of sustained ventricular tachycardia [1] and later for acute cardioversion of AF and for sinus rhythm maintenance. Currently, the use of flecainide in atrial fibrillation represents the main indication of the drug. Nevertheless, despite its effectiveness and safety profile it is still underused [2]
Pharmacokinetics
Flecainide, administered bis in die (immediate-release form) or once daily (controlled-release form), is nearly completely absorbed from the gastrointestinal tract with very high bioavailability (from 85% to 90%) [3]. Serum concentration (SC) ranging from 0.2 to 1 mcg/ml provide the greatest therapeutic benefit but higher concentrations are associated with proarrhythmic side effects [4]. The apparent volume of distribution is wide, with only about 40% of drug bound to plasma proteins [5]. Flecainide is metabolized in the liver via cytochrome (CYP2D6 and CYP1A2) in meta-O-dealkylated flecainide (active, but about one-fifth as potent) and meta-O-dealkylated lactam (inactive form), then excreted in the urine. Since flecanide is mainly metabolized via cytochrome CYP2D6 [6], CYP2D6 inhibitors increase its plasmatic concentration and inductors decrease it. [7]. About 30% of an orally administered dose escapes liver metabolism and is excreted in the urine unchanged [3]. Elimination half-life is about 20 hours (range: 12-27 hours) [8], unaffected by dose, but it may be prolonged until 70 hours in patients with heart failure, renal disease (creatinine clearance < 50 ml/min) and liver disease [8] (Table 1).
Table 1. Pharmacokinetics properties of flecainide.
|
|
Pharmacokinetics |
|
Absorption |
85-90% absorbed from the gastrointestinal tract; Serum concentration peak reached in 1-3 h. |
|
Distribution |
Wide volume of distribution; 40% binded to plasma proteins. |
|
Metabolism |
30% escapes liver metabolism. |
|
Excretion |
Excreted by urine; Elimination half-life: about 20 hours. |
Pharmacodynamics
Flecainide works blocking the open-state fast inward Na+ channel Nav 1.5 [8] in a rate- and voltage-dependent manner, reducing the maximum rate of phase 0 rise of the action potential (Vmax) in fast channel-dependent myocardial fibers (mostly in His-Purkinje tissue and ventricular muscle, followed by atrial muscle) [9]. Moreover, flecainide at low dose inhibits rapid component of the delayed rectifier K+ current (IKr) [10] [11] while at higher concentrations inhibits other K+ channels (Kito) [12]. Flecainide exerts a variable effect in terms of action potential duration (APD) and effective refractory period (ERP) on ventricular fibers and the Purkinje fibers. In fact, while the ERP and APD in atrial and ventricular fibers are prolonged, in the Purkinje system are shortened [13]. This effect is probably consistent with Na+ channel blockade [8][11][14]). Flecainide also inhibits ryanodine receptor 2 (RyR2) reducing calcium sparks [4] and arrhythmogenic calcium currents [15]. Moreover, flecainide reduces Na+ and Ca2+ inflow in myocardial cells and exerts a negative inotropic effect. Intravenous administration reduces cardiac output and stroke volume [16][17]especially for patients with coronary artery disease [4][18][19][20] or left ventricular (LV) dysfunction [20] while has minimal hemodynamic effects in patients with normal, or nearly normal, ventricular function [4][8][21][22]
Furthermore, flecainide reduces atrial remodeling and oxidative stress by inhibiting intracellular Ca2+ accumulation. Indeed, atrial activation, when caused by atrial fibrillation, rapidly leads to intracellular calcium accumulation due to Na+/Ca2+ exchanger. This results in myocardial remodeling and mitochondrial dysfunction related to oxidative stress [23]
2. Vademecum for the Management of Flecainide
A 12-lead ECG is mandatory before starting the therapy with flecainide and after 1-2 weeks after its administration; symptomatic bradycardia, second degree or superior atrioventricular (AV) block, QRS duration longer than 120 ms or Brugada syndrome contraindicate the prescription of flecainide. [24] [7]
It is reasonable to perform an echocardiogram to evaluate LV function and exercise stress testing in high-risk patients to exclude the possibility of coronary artery disease. It is strongly suggested to test the first dose under medical observation. The minimum effective plasma concentration of flecainide is about 200 ng/mL while optimal range is between 200 ng/mL and 400 ng/mL [25]. This plasma concentration leads to a QRS prolongation of about 10 ms; a prolongation of 40 ms or more is associated to an increased probability of cardiovascular adverse effects [25]. A practical approach to flecainide dose ranging, in absence of kidney failure, is as follows [24] (see also—Figure 1):
- Exclude contraindications (structural heart disease, symptomatic bradycardia, second degree or superior AV block, QRS > 120 ms or Brugada syndrome).
- Record an ECG with a paper speed of 50 mm/sec and calculate the QRS duration (1 mm = 20 ms).
- Administer a loading oral dose of 250 mg (200 mg if the weight is lower than 70 kg).
- At plasma concentration peak, after 90–120 min, evaluate blood pressure and record an ECG with a paper speed of 50 mm/s and calculate the QRS duration.
- Rule out Brugada ECG pattern and AV block.
- If the QRS duration is increased within 20 ms, prescribe 100 mg twice daily or 200 mg once daily. Check again the ECG after one week.
- If the QRS duration is increased between 20 and 40 ms or is wider than 120 ms, prescribe 50 mg twice daily or 100 mg once daily. Check again the ECG after 5 days.
- If the QRS duration is increased more than 40 ms or is wider than 130 ms, or a Brugada pattern is detected, consider flecainide contraindicated in that patient
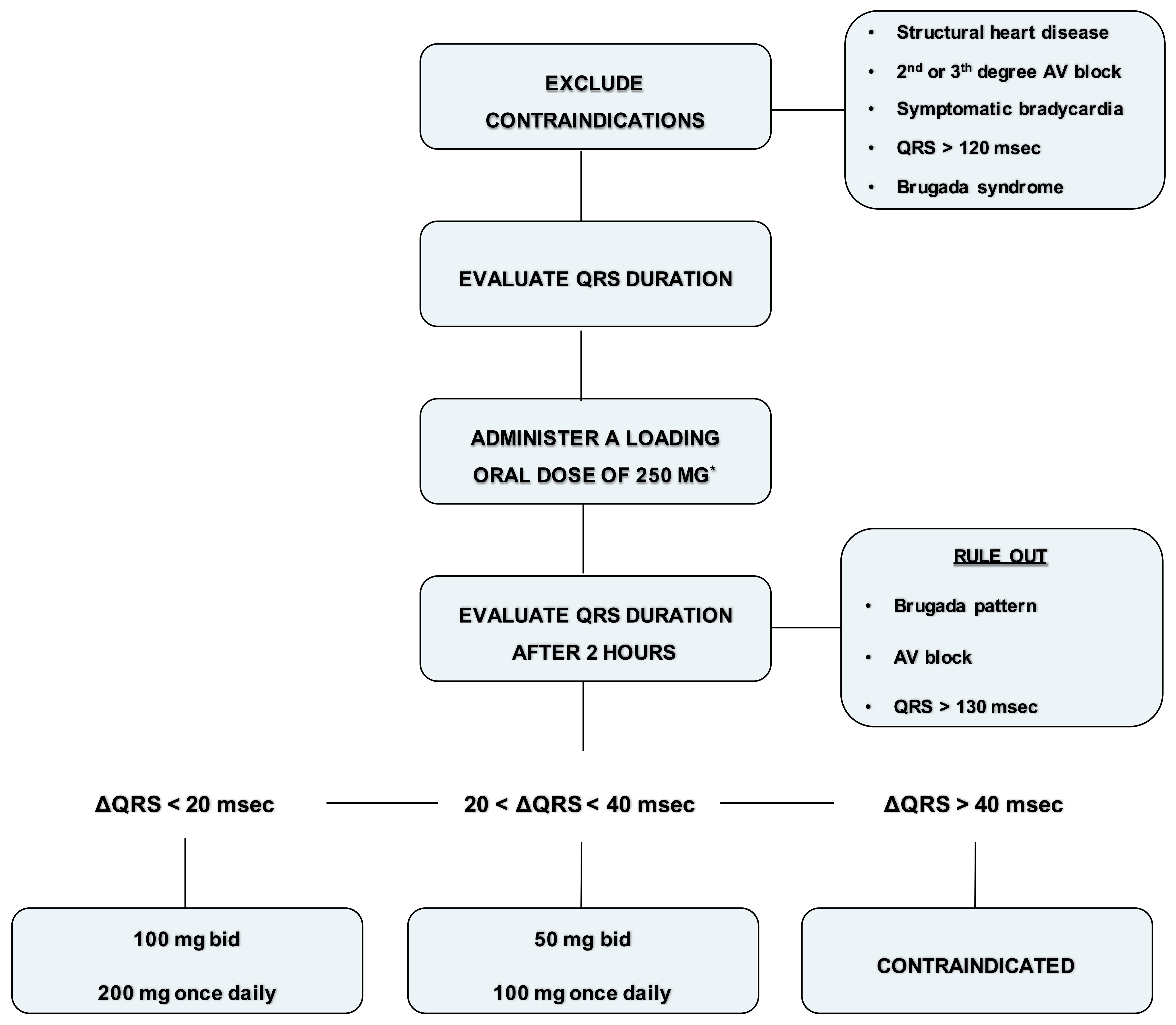
Figure 1. Flowchart for administration of flecainide. * Loading oral dose of 200 mg is recommended if the weight is lower than 70 kg.
There is a relationship between dose and concentration for both safety and efficacy. The incidence of adverse cardiovascular events rises with increasing flecainide plasma levels. However, the therapeutic effects, associated with greater than 90% suppression of premature ventricular beats (VPBs), are achieved at the range between 250–500 ng/mL of flecainide plasma levels. [25][26]. Clinicians should consider this aspect and adjust dosing according to the clinical response. [25]
Special Population
Periodic monitoring of flecainide plasma levels, blood pressure and ECG is required in patients with severe hepatic disease or severe renal failure. [25] [27]. Flecainide is predominantly excreted in the urine and the plasma elimination half-life is significantly prolonged in patients with renal impairment. [25][26][27]
Flecainide plasma levels and ECG monitoring are also indicated in elderly patients, who present decrease in renal function and an increased drug-drug interaction risk for the presence of comorbidities. [25] Furthermore, it has been demonstrated that there is an age-related decline of flecainide clearance in this population due to a CYP2D6 genotype. [6]
In pediatric patients a concentration of flecainide lower than the therapeutic concentration for adult patients is enough (concentration between 0.2 to 1 mcg/ml). [27]
3. Indication
Flecainide is an IC antiarrhythmic drug approved in 1984 from Food and Drug Administration for the suppression of sustained ventricular tachycardia and later for acute cardioversion of atrial fibrillation and for sinus rhythm maintenance [1]. Flecainide is used in paroxysmal supraventricular tachycardia (PSVT’s), including atrioventricular nodal re-entrant tachycardia (AVNRT), AV re-entrant tachycardia (AVRT) and atrial fibrillation/atrial flutter in patients who do not have structural heart disease. Flecainide is also an option in treating life-threatening ventricular arrhythmias. The use of flecainide in atrial fibrillation represents the main indication of the drug.
3.1. Flecainide in Atrial Fibrillation
in converting recent onset of atrial fibrillation
Flecainide is very effective in restoring sinus rhythm in acute setting [28] with high percentages of success, greater than both propafenone and amiodarone [29] as well as with shorter cardioversion times (50% of patients are actually cardioverted in 1h if intravenous administration and in 3 h if orally given) [30][31][32][33][34][35]
Flecainide is effective even when administered orally at a charge dose (“pill in the pocket strategy”). The "pill in the pocket" strategy is currently indicated as a therapeutic strategy in selected patients (class IIa, level of evidence B), with recent onset of atrial fibrillation without significant structural or ischemic heart disease, but only following efficacy and safety assessment [7]
in long term rhythm control
Flecainide is currently recommended as a first-line long-term rhythm control in atrial fibrillation in class of recommendation I and level of evidence A, in patients with normal left ventricular function and without structural heart disease [7]
The maintenance of the sinus rhythm is more advantageous than rate-control both in terms of survival [36] and quality of life [37][38]. Several studies showed that flecainide is safe and effective in reducing the recurrences of paroxysmal AF and the time between episodes when compared to other antiarrhythmic drugs (table 2)
Table 2. Flecainide for maintenance of sinus rhythm.
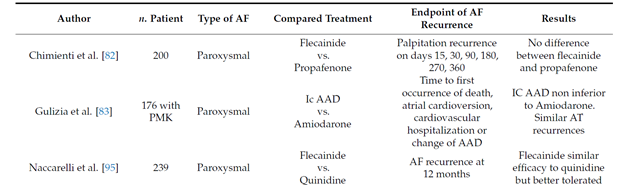

Long term oral flecainide administration reduce the incidence of atrial fibrillation recurrence in patients with paroxysmal and persistent atrial fibrillation, extending the time between two episodes [39][40][41][42][43]. A large study conducted on 994 patients with paroxysmal atrial fibrillation (PAF) showed that, after 9 months of treatment with flecainide at the mean dose of 200 mg, 65% patients were free from arrhythmia [44].
Recently, a retrospective study involving 144 patients with atrial fibrillation demonstrated that flecainide is effective and well tolerated, even at 12 months. After 6 and 12 months of treatment, 70.8% and 61.8% of patients respectively were symptomatically controlled [45].
3.2. Flecainide in Supraventricular Tachyarrhythmias
Flecainide is recommended for focal atrial tachycardia (if other treatment failed), in atrial flutter, in atrioventricular nodal re-entrant tachycardia (AVNRT) and atrioventricular re-entrant tachycardia (AVRT). In pregnant women should be considered for prevention of supraventricular tachycardia (SVT) in patients with Wolff–Parkinson–White (WPW) syndrome and without ischemic or structural heart disease. [46]
Flecainide reduces the recurrences of supraventricular arrhythmias, and this could be related to the inhibition of the potassium and calcium currents with consecutive reduction of cellular excitability. Usually, betablockers are used as a first line therapy in atrioventricular nodal re-entry tachycardia, but flecainide represents an alternative if beta-blockers are not effective or contraindicated. [27]
3.3. Flecainide in Ventricular Tachycardias
The treatment of ventricular tachycardia represented the first historical clinical indication for flecainide [27] [47]
Usually, ventricular tachycardias with a low heart rate are provoked by very slow conduction isthmuses and, on areas already characterized by very slow conduction velocity, flecainide is effective. [48]
Flecainide plays a key role in management of Premature Ventricular Complexes (PVC) and of Ventricular Tachycardia (VT) in structurally normal hearts with a class I indication and should be utilized as first line therapy in these patients beside radiofrequency ablation. Choice between drug therapy and ablation is based on patient choice and procedure complexity and risks.
Flecainide may be useful as add-on therapy in management of some rare channelopathies such as Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT) and some subtypes of long QT syndrome, LQTS type 3 and Andersen – Tawil Syndrome (LQTS type 7). [49]
Flecainide is very effective in CPVT which usually has a high frequency. In this arrhythmia, however, the efficacy seems to be related to a different mechanism of action. CPVT is induced by calcium overload due to sympathetic activation and to a diastolic calcium release from the sarcoplasmic reticulum through defective leaking RYR2 channels. Flecainide blocks the RYR2 channels allowing direct targeting of the molecular defect [50].
Flecainide is effective in patients with Long QT Syndrome (LQT) type 3 as it inhibits not only the peak but also the late component of the sodium current, inducing an abbreviation of the QT interval. This form, indeed, is provoked by mutations that increase the late sodium current [51]
3.4. Flecainide in Association with Other Antiarrhythmic Drugs
In some AF patients may be difficult to maintain sinus rhythm and symptomatic AF episodes remains a difficult therapeutic problem. Therefore, a combined anti-arrhythmic strategy may be necessary in some cases. Flecainide combination with amiodarone is interesting, not only because it may be effective when the efficacy of each is inadequate as a single-drug therapy, but also because it is relatively safe and may allow a reduction in their respective dosages and side effects [52] [53][54]
Beta blockers are the most used antiarrhythmic drug in association with flecainide. Capucci and colleagues have demonstrated that combination therapy with flecainide and metoprolol significantly reduced recurrences at 1-year follow-up when compared with flecainide alone in the whole population and in patients with persistent AF, while in patients with paroxysmal AF no benefit was observed over IC anti-arrhythmic drug-only. [55]
The association of flecainide with other AV blockers as digoxin is frequent; close monitoring of serum digoxin concentrations is recommended because it could increase 6 hours after co-administration with flecainide by 15–19% [56][57]
In the same way, it is necessary to consider with caution the combination of flecainide with verapamil, because of potential additive effects on myocardial contractility and on atrioventricular conduction [57]
3.5. Contraindications
According to 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation, flecainide is contraindicated in patients with ischemic heart disease and/or significant structural heart disease. Flecainide may induce hypotension and mild QRS complex widening. Furthermore, may increase atrial flutter cycle length, thus promoting 1:1 atrioventricular conduction and increasing ventricular rate; this risk can be reduced by concomitant administration of an atrioventricular nodal blocking drug such as a beta-blocker or non-dihydropyridinecalcium-channel blocker[7]
A practical guide on the management of adverse events due to flecainide is provided in Table 3.
Table 3. Management of adverse events due to flecainide.
| Adverse Event | Incidence | Indication |
|---|---|---|
| Drug induced Brugada | <1% | Discontinue |
| QRS increased more than 40 ms or wider than 130 ms | <1% | Discontinue |
| QRS increased more than 20 ms | 1–2% | Reduce Dosage |
| Bradyarrhythmia, sinus pause, AV block | 1–2% | Discontinue |
| Hypotension | 3–5% (mostly with IV) | Reduce Dosage |
| 1:1 atrial flutter | 3–5% | Discontinue and consider ablate CTI dependent-flutter |
| Worsening heart failure | <1% | Discontinue |
| Extracardiac effects (dizziness, tremor, nausea) | 1–2% | Reduce Dosage |
AV: atrioventricular; CTI: cavotricuspid isthmus; IV: intravenous.
4. Controlled release flecainide
The controlled-release (CR) formulation of flecainide acetate allows once-a-day administration therapy. The pharmacokinetic profile is characterized by a reduced and delayed reaching maximum concentration (Cmax) and lower fluctuations of plasma concentrations during a dosing interval compared with immediate-release form. Serum concentration peak of slow-release form is reached in 26 h, the steady state plasma level is reached after 4-5 days ranging from 270 to 330 ng/ml far from plasma level at risk of side effects [58]. After repeated-dose administration, maximum and minimum QRS prolongation does not differ between the two forms but with the CR-formulation there is a less variability compared with the immediate-release form [58]. Moreover, CR formulation improves treatment compliance and, reducing variability and peaks, decreases the risk of side effects and interactions with other drugs, preserving clinical benefit. Controlled-release flecainide is also effective in long-term prevention of atrial fibrillation recurrences. In 227 outpatients with paroxysmal atrial fibrillation and treated for 24 weeks with controlled-release flecainide at the dose of 200 mg, there was a decrease of incidence of paroxysmal atrial fibrillation episodes from 28.6% at baseline to 11% at the end of the study (p<0.0001) recording by Holter [59].
Recently, in a prospective multicenter observational study that enrolled 679 patients with paroxysmal and persistent atrial fibrillation, was reported that a treatment with flecainide at controlled-release form improve quality of life and have a good safety profile. Indeed, the treatment with CR flecainide reduced the CCS-SAF score from 1.64 at baseline to 1.32 at the end of the study (p < 0.0001) and only the 0,6% of patients experienced non serious adverse events (dizziness, presyncope, flushing and a new-onset left bundle branch block). Furthermore, the compliance to treatment with flecainide at controlled-release form was elevated during 12-weeks period (100% in 93,6% of patients). This high compliance may reflect the once daily dosing regimen and this trend has a considerable impact on cardiovascular disease medications [60].
Moreover, patients in treatment with flecainde immediate release form could be switched to the controlled-release formulation on a “mg-for-mg” daily dose basis, without any special need to monitor serum level. Harrison and colleagues demonstrated that all the subject in treatment with immediate release form of flecainide were successfully switched to the controlled-release form with this approach. [61]
5. Conclusions
Flecainide, administered bis in die (immediate-release form) or once daily (controlled-release form), is effective for the acute termination and for the chronic suppression of atrial fibrillation and ventricular arrhythmia. An excellent safety profile is described in patients with minimal or no signs of structural heart disease while mounting promising evidence will be available in patient with cardiomyopathy. A 12-lead ECG is required before starting therapy while ECG monitoring is suggested in case of drug adjustments or concomitant therapy with other antiarrhythmic drugs, particularly in the elderly and in patients with hepatic and/or renal dysfunction.
Despite the evidence supporting the efficacy and tolerability profile of flecainide, it is still underused due to the wrong idea about the risk for ventricular proarrhythmia. This aspect is even more clear with the controlled release form.
The immediate-release form (IR) and controlled-release form (CR) of flecainide have the same pharmacokinetic properties (absorption, distribution and excretion), but the pharmacokinetic profile of CR flecainide is characterized by a reduced and delayed reaching maximum concentration (Cmax) and lower fluctuations of plasma concentrations during a dosing interval compared with immediate-release form. Controlled release formulation improves treatment compliance and, reducing variability and peaks, decreases the risk of side effects and interactions with other drugs preserving clinical benefit.
This entry is adapted from the peer-reviewed paper 10.3390/jcm10071456
References
- Hoffmeister HM, Hörmann HP, Beyer M, Seipel L.; Negative inotropic effect of flecainide on the post-ischemic heart with limited contractile function.. Z Kardiol 1990, 79, 189-92, .
- Nieuwlaat R, Capucci A, Camm AJ, et al.; Atrial fibrillation management: a prospective survey in ESC member countries: the Euro Heart Survey on Atrial Fibrillation. . Eur Heart J 2005, 26, 2422-34, 10.1093/eurheartj/ehi505.
- Conard GJ, Ober RE.; Metabolism of flecainide. . Am J Cardiol 1984, 53, 41-51, 10.1016/0002-9149(84)90501-0.
- Anderson, J.L.; Stewart, J.R.; Perry, B.A.; Van Hamersveld, D.D.; Johnson, T.A.; Conard, G.J.; Chang, S.F.; Kvam, D.C.; Pitt, B.; Oral Flecainide Acetate for the Treatment of Ventricular Arrhythmias.. N. Engl. J. Med. 1981, 305, 473–477, 10.1056/NEJM198108273050901.
- Tjandra-Maga TB, Verbesselt R, Van Hecken A, Mullie A, De Schepper PJ.; Flecainide: single and multiple oral dose kinetics, absolute bioavailability and effect of food and antacid in man.. Br J Clin Pharmacol 1986, 22, 309-16, 10.1111/j.1365-2125.1986.tb02892.x.
- Doki K, Homma M, Kuga K, Aonuma K, Kohda Y; CYP2D6 genotype affects age-related decline in flecainide clearance: a population pharmacokinetic analysis.. Pharmacogenet Genomics 2012, 22, 777–783, 10.1097/FPC.0b013e3283588fe5.
- Hindricks, Potpara, Dagres, Arbelo, Bax, Blomstrom-Lundqvist, Boriani, Castella et al.; 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS). European Heart Journal 2020, 42, 373-498, 10.1093/eurheartj/ehaa612 .
- Holmes B, Heel RC.; Flecainide. A preliminary review of its pharmacodynamic properties and therapeutic efficacy. . Drugs 1985, 29, 1-33, 10.2165/00003495-198529010-00001.
- Josephson MA, Ikeda N, Singh BN.; Effects of flecainide on ventricular function: clinical and experimental correlations.. Am J Cardiol 1984, 53, 95-100, 10.1016/0002-9149(84)90510-1.
- Anno T, Hondeghem LM.; Interactions of flecainide with guinea pig cardiac sodium channels. Importance of activation unblocking to the voltage dependence of recovery. . Circ Res 1990, 66, 789-803, 10.1161/01.res.66.3.789.
- Follmer CH, Colatsky TJ.; Block of delayed rectifier potassium current, IK, by flecainide and E-4031 in cat ventricular myocytes.. Circulation 1990, 82, 289-93, 10.1161/01.cir.82.1.289.
- Tamargo J, Caballero R, Gómez R, Valenzuela C, Delpón E.; Pharmacology of cardiac potassium channels. . Cardiovasc Res 2004, 62, 9-33, 10.1016/j.cardiores.2003.12.026.
- Singh BN, Nademanee K, Josephson MA, Ikeda N, Venkatesh N, Kannan R.; The electrophysiology and pharmacology of verapamil, flecainide, and amiodarone: correlations with clinical effects and antiarrhythmic actions.. Ann N Y Acad Sci 1984, 432, 210-35, 10.1111/j.1749-6632.1984.tb14522.x.
- Campbell TJ, Vaughan Williams EM.; Voltage- and time-dependent depression of maximum rate of depolarisation of guinea pig ventricular action potentials by two new antiarrhythmic drugs, flecainide and lorcainide. . Cardiovasc Res 1983, 17, 251-8, 10.1093/cvr/17.5.251.
- Mehra D, Imtiaz MS, van Helden DF, Knollmann BC, Laver DR.; Multiple modes of ryanodine receptor 2 inhibition by flecainide.. Mol Pharmacol 2014, 86, 696-706, 10.1124/mol.114.094623.
- Muhiddin KA, Turner P, Blackett A.; Effect of flecainide on cardiac output.. Clin Pharmacol Ther 1985, 37, 260-3, 10.1038/clpt.1985.37.
- Legrand V, Vandormael M, Collignon P, Kulbertus HE.; Hemodynamic effects of a new antiarrhythmic agent, flecainide (R-818), in coronary heart disease. Am J Cardiol 1983, 51, 422-6, 10.1016/s0002-9149(83)80073-3.
- Duff HJ, Roden DM, Maffucci RJ, et al.; Suppression of resistant ventricular arrhythmias by twice daily dosing with flecainide.. Am J Cardiol 1981, 48, 1133-40, 10.1016/0002-9149(81)90331-3.
- Hodges M, Haugland JM, Granrud G, et al.; . Suppression of ventricular ectopic depolarizations by flecainide acetate, a new antiarrhythmic agent.. Circulation 1982, 65, 879-85, 10.1161/01.cir.65.5.879.
- Josephson MA, Kaul S, Hopkins J, Kvam D, Singh BN.; Hemodynamic effects of intravenous flecainide relative to the level of ventricular function in patients with coronary artery disease. . Am Heart J 1985, 109, 41-5, 10.1016/0002-8703(85)90413-2. .
- De Paola AA, Horowitz LN, Morganroth J, et al.; Influence of left ventricular dysfunction on flecainide therapy. . J Am Coll Cardiol 1987, 9, 163-8, 10.1016/s0735-1097(87)80096-7.
- Le Grand B, Le Heuzey JY, Perier P, et al.; Cellular electrophysiological effects of flecainide on human atrial fibres. . Cardiovasc Res 1990, 24, 232-8, 10.1093/cvr/24.3.232.
- Iwai T, Tanonaka K, Inoue R, Kasahara S, Motegi K, Nagaya S, Takeo S.; Sodium accumulation during ischemia induces mitochondrial damage in perfused rat hearts. . Cardiovasc Res. 2002, 55(1), 141-9, 10.1016/s0008-6363(02)00282-1.
- Lavalle, Magnocavallo, Straito, Santini, Forleo, Grimaldi, Badagliacca, Lanata and Ricci.; . Flecainide How and When: A Practical Guide in Supraventricular Arrhythmias.. J. Clin. Med. 2021, 10, 1456, 10.3390/jcm10071456.
- Tamargo J, Le Heuzey JY, Mabo P.; Narrow therapeutic index drugs: a clinical pharmacological consideration to flecainide.. Eur J Clin Pharmacol 2015, 71(5), 549-67, 10.1007/s00228-015-1832-0.
- Salerno DM, Granrud G, Sharkey P et al; Pharmacodynamics and side effects of flecainide acetate. . Clin Pharmacol Ther 1986, 40, 101–107, 10.1038/clpt.1986.145.
- Paolini, Stornati, Guerra, Capucci.; Flecainide: Electrophysiological properties, clinical indications, and practical aspects. . Pharmacological Research 2019, 148, 104443, 10.1016/j.phrs.2019.104443.
- McNamara RL, Bass EB, Miller MR, et al.; Management of new onset atrial fibrillation. . Evid Rep Technol Assess (Summ) 2000, 12, 1-7, .
- Martínez-Marcos FJ, García-Garmendia JL, Ortega-Carpio A, Fernández-Gómez JM, Santos JM, Camacho C.; Comparison of intravenous flecainide, propafenone, and amiodarone for conversion of acute atrial fibrillation to sinus rhythm. . Am J Cardiol 2000, 86, 950-3, 10.1016/s0002-9149(00)01128-0.
- Boriani G, Biffi M, Capucci A, et al.; Oral propafenone to convert recent-onset atrial fibrillation in patients with and without underlying heart disease.A randomized, controlled trial. . Ann Intern Med 1997, 126, 621-5, 10.7326/0003-4819-126-8-199704150-00006.
- Capucci A, Boriani G, Botto GL, et al.; Conversion of recent-onset atrial fibrillation by a single oral loading dose of propafenone or flecainide. . Am J Cardiol 1994, 74, 503-5, 10.1016/0002-9149(94)90915-6.
- Crijns HJ, van Wijk LM, van Gilst WH, Kingma JH, van Gelder IC, Lie KI.; Acute conversion of atrial fibrillation to sinus rhythm: clinical efficacy of flecainide acetate. Comparison of two regimens. . Eur Heart J 1988, 9, 634-8, 10.1093/oxfordjournals.eurheartj.a062553.
- Boriani G, Biffi M, Capucci A, et al.; Conversion of recent-onset atrial fibrillation to sinus rhythm: effects of different drug protocols.. Pacing Clin Electrophysiol 1998, 21, 2470-4, 10.1111/j.1540-8159.1998.tb01203.x.
- Capucci A, Lenzi T, Boriani G, et al.; Effectiveness of loading oral flecainide for converting recent-onset atrial fibrillation to sinus rhythm in patients without organic heart disease or with only systemic hypertension. . Am J Cardiol 1992, 70, 69-72, 10.1016/0002-9149(92)91392-h.
- Romano S, Fattore L, Toscano G, et al.; Effectiveness and side effects of the treatment with propafenone and flecainide for recent-onset atrial fibrillation. . Ital Heart J Suppl 2001, 2, 41-5, .
- Alboni, P.; Paparella, N.; Cappato, R.; Candini, G.C.; Direct and Autonomically Mediated Effects of Oral Flecainide. . Am. J. Cardiol 1988, 61, 759–763, 10.1016/0002-9149(88)91062-4.
- Hellestrand, K.J.; Nathan, A.W.; Bexton, R.S.; Camm, A.J.; Electrophysiologic Effects of Flecainide Acetate on Sinus Node Function,Anomalous Atrioventricular Connections, and Pacemaker Thresholds. . Am. J. Cardiol. 1984, 53, 30B-38B, 10.1016/0002-9149(84)90499-5.
- Anderson, J.L.; Lutz, J.R.; Allison, S.B.; Electrophysiologic and Antiarrhythmic Effects of Oral Flecainide in Patients with InducibleVentricular Tachycardia. J. Am. Coll. Cardiol. 1983, 2, 105–114, 10.1016/s0735-1097(83)80382-9.
- Anderson JL, Gilbert EM, Alpert BL, Henthorn RW, Waldo AL, Bhandari AK, Hawkinson RW, Pritchett EL.; Prevention of symptomatic recurrences of paroxysmal atrial fibrillation in patients initially tolerating antiarrhythmic therapy. A multicenter, double-blind, crossover study of flecainide and placebo with transtelephonic monitoring. Flecainide supraventricular tachycardia study group. . Circulation 1989, 80, 1557–1570, 10.1161/01.cir.80.6.1557.
- Pietersen AH, Hellemann H.; Usefulness of flecainide for prevention of paroxysmal atrial fibrillation and flutter. Danish-Norwegian flecainide multicenter study group. . Am J Cardiol 1991, 67, 713–717, 0.1016/0002-9149(91)90527-r.
- Van Gelder IC, Crijns HJ, Van Gilst WH, Van Wijk LM, Hamer HP, Lie KI.; Efficacy and safety of flecainide acetate in the maintenance of sinus rhythm after electrical cardioversion of chronic atrial fibrillation or atrial flutter. Am J Cardiol 1989, 64, 1317–1321, .
- Kirchhof P, Andresen D, Bosch R, Borggrefe M, Meinertz T, Parade U, Ravens U, Samol A, Steinbeck G, Treszl A, Wegscheider K, Breithardt G.; Short-term versus long-term antiarrhythmic drug treatment after cardioversion of atrial fibrillation (FLec-SL): a prospective, randomised, open-label, blinded endpoint assessment trial.. Lancet 2012, 380(9838), 238-46, 10.1016/S0140-6736(12)60570-4.
- Echt and Ruskin.; Use of Flecainide for the Treatment of Atrial Fibrillation.. M Am J Cardiol 2020, 00, 1−11, 10.1016/j.amjcard.2019.12.041.
- Clementy J, Dulhoste MN, Laiter C, Denjoy I, Dos Santos P; Flecainide acetate in the prevention of paroxysmal atrial fibrillation: a nine-month follow-up of more than 500 patients. Am J Cardiol 1992, 70, 44A-49A, 10.1016/0002-9149(92)91077-h.
- Muzzey, Tellor , Ramaswamy, Schwarze and Armbruster.; Flecainide is well-tolerated and effective in patient with atrial fibrillation at 12 months:a retrospective study. . Ther Adv Cardiovasc Dis 2020, 14, 1–7, 10.1177/1753944720926824.
- Brugada, Katritsis, Arbelo, Arribas, Bax et al.; 2019 ESC Guidelines for the management of patients with supraventricular tachycardia.. European Heart Journal 2020, 41, 655-720, 10.1093/eurheartj/ehz467.
- Hoffmeister, Hörmann, Beyer, Seipel.; [Negative inotropic effect of flecainide on the post-ischemic heart with limited contractile function].. Z Kardiol. 1990, 79(3), 189-92, .
- Lavalle, Trivigno, Vetta, Magnocavallo, Mariani, Santini, Forleo, Grimaldi, Badagliacca, Lanata and Ricci.; Flecainide in Ventricular Arrhythmias: From Old Myths to New Perspectives. . J. Clin. Med 2021, 10, 3696, 10.3390/jcm10163696.
- Priori, Blomstro ̈m-Lundqvist, Mazzanti, Bloma, Borggrefe, Camm, Elliott et al.; 2015 ESC Guidelines for the managementof patients with ventricular arrhythmiasand the prevention of sudden cardiac death. . European Heart Journal 2015, 36, 2793–2867, 10.1093/eurheartj/ehv316.
- Hilliard FA, Steele DS, Laver D, et al.; Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass. . Journal of Molecular and Cellular Cardiology 2010, 48, 293–301, 10.1016/j.yjmcc.2009.10.005.
- Belardinelli L, Giles WR, Rajamani S, Karagueuzian HS, Shryock JC.; Cardiac late Na+ current: Proarrhythmic effects, roles in long QT syndromes, and pathological relationship to CaMKII and oxidative stress. . Heart Rhythm 2015, 12, 440–448, 10.1016/j.hrthm.2014.11.009.
- Mary-Rabine, L.; Telerman, M.; Long Term Evaluation of Flecainide Acetate in Supraventricular Tachyarrhythmias. . Acta Cardiol. 1988, 43, 37–48, .
- Shea, P.; Lal, R.; Kim, S.S.; Schechtman, K.; Ruffy, R.; Flecainide and Amiodarone Interaction. J. Am. Coll. Cardiol 1986, 7, 1127–1130, 10.1016/s0735-1097(86)80234-0.
- Coumel, P.; Chouty, F.; Slama, R.; Logic and Empiricism in the Selection of Antiarrhythmic Agents: The Role of Drug Combinations.. Drugs 1985, 29, 68–76, 10.2165/00003495-198500294-00014.
- Capucci, A.; Piangerelli, L.; Ricciotti, J.; Gabrielli, D.; Guerra, F.; Flecainide–Metoprolol Combination Reduces Atrial Fibrillation Clinical Recurrences and Improves Tolerability at 1-Year Follow-up in Persistent Symptomatic Atrial Fibrillation. . Europace 2016, 18, 1698–1704, 10.1093/europace/euv462.
- Trujillo, T.C.; Nolan, P.E.; Antiarrhythmic Agents: Drug Interactions of Clinical Significance. . Drug Saf. 2000, 23, 509–532, 10.2165/00002018-200023060-00003.
- Holtzman, J.L.; Finley, D.; Mottonen, L.; Berry, D.A.; Ekholm, B.P.; Kvam, D.C.; McQuinn, R.L.; Miller, A.M.; The Pharmacodynamic and Pharmacokinetic Interaction between Single Doses of Flecainide Acetate and Verapamil: Effects on Cardiac Function and Drug Clearance. Clin. Pharm. 1989, 46, 26–32, 10.1038/clpt.1989.102.
- Tennezé L, Tarral E, Ducloux N, Funck-Brentano C.; Pharmacokinetics and electrocardiographic effects of a new controlled-release form of flecainide acetate: comparison with the standard form and influence of the CYP2D6 polymorphism. . Clin Pharmacol Ther 2002, 72, 112-22, 10.1067/mcp.2002.125946.
- Aliot E, De Roy L, Capucci A, et al.; Safety of a controlled-release flecainide acetate formulation in the prevention of paroxysmal atrial fibrillation in outpatients. Ann Cardiol 2003, 52, 34-40, 10.1016/s0003-3928(02)00183-x.
- Tzeis S, Tsiachris D, Asvestas D, Kourouklis S, Patsourakos F, Karlis D, Kouskos G, Papadimitriou G, Gavriilidou M, Vatkalis N, Kapetanios K, Koufaki P, Taxiarchou E, Giannakoulas G; Beneficial Effect of Flecainide Controlled Release on the Quality of Life of Patients with Atrial Fibrillation-the REFLEC-CR Study.. Cardiovasc Drugs Ther 2020, 34(3), 383-389, 10.1007/s10557-020-06971-5.
- Harrison, Chang, Dahms, Miller, Fox, Donnell.; Multiple dose pharmacokinetics of flecainide acetate from a once-a-day formulation in humans. European journal of clinical research 1994, 5, 151‐160, .