Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemical Research Methods
A convenient method for the synthesis of the first generation PAMAM dendrimers based on the thiacalix[4]arene has been developed for the first time. Three new PAMAM-calix-dendrimers with the macrocyclic core in cone, partial cone, and 1,3-alternate conformations were obtained with high yields.
- thiacalixarene
- DNA
- dendrimer
- PAMAM
1. Introduction
In recent years, research aimed at developing DNA vaccines has shown great potential [1]. These medicinal products are already successfully used in veterinary medicine [2]. Recently, Inovio Pharmaceuticals Corporation (Plymouth Meeting, PA) proposed a DNA vaccine for the prevention of SARS COVID-19 [3]. A significant increase in research on the use of DNA-based drugs for the treatment of other diseases has also been noted in recent years [4]. Wound-healing, anti-inflammatory, and antithrombotic drugs have been clinically tested and are now commercially available. For example, Jazz Pharmaceuticals proposed the drug Defitelio®® (defibrotide sodium), which is the oligonucleotide obtained by depolymerization of porcine intestinal mucosal DNA for the treatment of hepatic veno-occlusive disease [5]. In addition, polynucleotides obtained from marine fish exert a wide therapeutic use [6,7,8].
The area of scientific research devoted to the development of DNA storage systems has become important due to the widespread application of DNA macromolecules [9]. Preservation of nucleic acid-based drugs from enzymatic degradation in the bloodstream is considered an important point [10]. The necessity of the storing of biomolecules is not limited to disease therapy. Intensive research has been carried out recently into preserving genetic material long-term to establish kinship and store information [11,12]. Among the methods of DNA storing, freezing is of great importance. However, chemical encapsulation is under intensive study as well. It is well known that DNA is able to bind with small molecules in three different ways, i.e., by electrostatic interactions, intercalation, or groove binding. All of these are important in different applications [13,14,15]. It is obvious that the structure of DNA can be preserved most closely to the native one during binding via electrostatic interactions; therefore, this is the most convenient method for DNA storing.
Currently, synthetic non-viral DNA delivery and storage systems are intensively developing. Dendrimers capable of forming dendriplexes with DNA are the most promising variants of the synthetic vectors for DNA. Their ability to pH-responsive osmotic swelling and endosomal membrane rupture is undoubtedly important for the transfection of genetic material [10,16]. The search for new dendrimeric compounds with fewer disadvantages is an important area of synthetic chemistry. Macrocyclic compounds such as cyclodextrins, pillararenes, and the (thia)calixarenes with positively charged groups are promising for use as a dendrimer core for binding DNA as a biological polyanion [17,18,19]. We used thiacalix[4]arene as a core of dendrimer compounds. (Thia)calixarene scaffold is non-toxic. It is known that the covalent binding of various toxic molecules with (thia)calixarene scaffold decreases their toxicity [20,21]. The possibility of easy synthesis of stereoisomers with different spatial arrangements of substituents is another advantage of the thiacalixarene platform. Relatively rigid spatial fixation of binding groups provides high selectivity of guest molecule binding [22,23,24]. It should be noted that the thiacalixarene scaffold exerts a multivalent binding effect [25,26] by fixing binding sites in space, which is very important in the case of DNA [27]. The use of thiacalix[4]arene in cone, partial cone, and 1,3-alternate conformation will lead to different shapes of the resulting dendrimers. True symmetric dendrimer shape in case of the core with the macrocycle in 1,3-alternate conformation, as well as asymmetrical shape [10] in case of those in cone and partial cone conformation, can be expected (Figure 1). In the dendrimer with the core in cone conformation (Figure 1), the hydrophilic dendritic part and lipophilic macrocyclic part will be strictly parted. The uniqueness of this structure is that it is amphiphilic, contrary to classical dendrimers. By using dendrimers of a different shape described above for the DNA binding, we will be able not only to achieve the multivalency effect of binding but also to obtain dendriplexes of different structures.
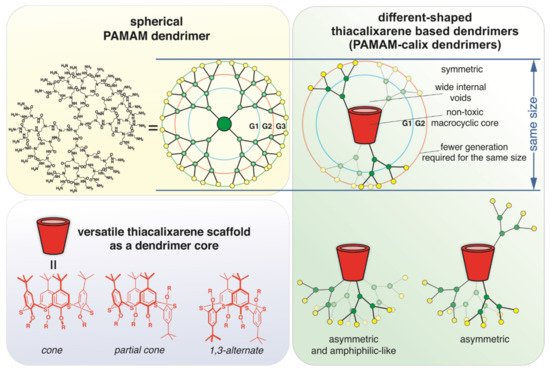
Figure 1. Schematic representation of the design of thiacalix[4]arene-based dendrimers.
2. Synthesis of (Poly)Amidoamine Dendrimers Based on the p-tert-Buthylthiacalix[4]arene (PAMAM-Calix-Dendrimer) in Different Conformations
Synthesis of the poly(amidoamine) (PAMAM) dendrimers is well described in the literature [28,29]. The divergent approach is the optimal one to their synthesis. It involves the use of repetitive sequences of reactions. First, the core containing primary amino groups is introduced into the Michael addition reaction with methyl acrylate for branching. Then, the obtained precursor (called half-generation) is involved in the reaction with an excess of the diamine. This leads to the formation of a full generation of the PAMAM dendrimer. Side reactions during the synthesis of dendrimers often cause defects in their structure. The literature [30,31] describes both the range of possible side reactions and the main methods for preventing their occurrence.
Despite a large number of advantages in the application of the PAMAM dendrimers [16,29], they have a number of significant disadvantages and limitations. The most significant is the increase of in vivo and in vitro cytotoxicity of dendrimers in the direction from lower to higher generation and, accordingly, the high toxicity of upper generations dendrimers [32]. Cytotoxicity of the high generation dendrimers is due to a large number of positive charges on their surface, which can induce morphological changes in cells or cause cell lysis by disrupting the integrity of the negatively charged phospholipid bilayers of the cell membranes [33]. The structure of dendrimeric molecules with classical core not capable of serious conformational changes is another limitation. Therefore, the shape of such dendrimer molecules is close to spherical. In addition, despite the fact that currently, PAMAM dendrimers are commercially available up to generation 10, the cost of upper generation (G > 3) dendrimers is high [34].
The size of thiacalix[4]arene [22] is comparable to that of the PAMAM dendrimers of lower generations with ethylenediamine (EDA) core [29]. The PAMAM dendrimers of 1st–3rd generation with thiacalix[4]arene core are assumed to be comparable in size with the dendrimers with the EDA core of higher generations. In addition, the interior void space of the thiacalix[4] arene-based dendrimer will be significantly larger. This allows obtaining dendrimers that can effectively bind biopolymers and other guest molecules using fewer steps of the synthesis.
Our approach combines the advantages of dendrimers and thiacalixarenes and reduces their disadvantages. It will make it possible to obtain low-toxic dendrimer macromolecules with a wide internal void that are capable of efficient multi-point interaction with substrates.
There are few examples of the synthesis of low generation dendrimers based on classical calix[4]arene in the literature [35,36]. They are limited by the use of calixarene in one spatial conformation, either cone or 1,3-alternate. It is also worth noting that there are no examples of the synthesis of high generation (G > 2) PAMAM dendrimers based on the thiacalix[4]arene platform.
In one recent publication [37], the authors described an improved approach to the synthesis and purification of the PAMAM dendrimers with a 1,4-diaminobutane core based on Tomalia’s previous works [29,38]. This approach allows to minimize the probability of side reactions and to obtain the PAMAM dendrimers in high yields. We have adapted this method to the synthesis of thiacalixarene-based dendrimers.
We have chosen the macrocyclic compounds G0 in cone (G0-cone), partial cone (G0-paco), and 1,3-alternate (G0-1,3-alt) conformation [26] as cores for the synthesis of the PAMAM dendrimers based on thiacalix[4]arene. Hereby they are named as PAMAM-calix-dendrimers. Starting compounds contain four primary amino groups in their structure. This makes it possible to effectively apply a divergent approach for the dendrimer synthesis (Scheme 1). In order to minimize the possibility of side reactions, the temperature did not exceed 30 °C at all stages of the synthesis, isolation, and purification.
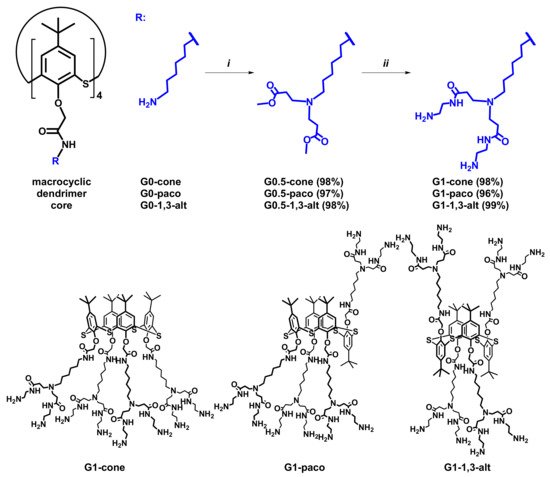
Scheme 1. Reagents and conditions: (i) methyl acrylate, methanol, r.t., 12 h; (ii) ethylenediamine, methanol, r.t., 70 h.
At the first stage of the synthesis, the compounds G0-cone, G0-paco, and G0-1,3-alt were involved in the reaction with methyl acrylate for branching creation. The reaction was carried out for 12 h at room temperature. As a result, the half-generation of PAMAM-calix-dendrimers, the compounds G0.5-cone, G0.5-paco, and G0.5-1,3-alt, were obtained with high yields (97–98%). Synthesis of the first generation dendrimer was the second stage. We used a large EDA excess (20 EDA equivalents per one ester group) to prevent side reactions of intra- and intermolecular crosslinking. Also, the complete removal of the EDA excess after the reaction is important in the synthesis of the PAMAM dendrimers. Otherwise, the residual EDA leads to trailing generation dendrimers due to the reaction between the EDA and methyl acrylate in the next stages. Taking these factors into account, a solution of the compounds G0.5-cone, G0.5-paco, and G0.5-1,3-alt in methanol was added to that of EDA in ice-cooled methanol. The reaction mixture was stirred for 70 h to ensure complete reaction of all the ester groups. Typically, azeotropic distillation in methanol:toluene (1:9) mixture was used to remove EDA from the reaction mixture completely [26,38]. After removal of the solvent and EDA from the reaction mixture, the residue was dissolved in methanol:toluene mixture, and all volatiles was removed under reduced pressure. This procedure was repeated seven times. Then the excessive toluene was removed by azeotropic distillation with methanol. As a result, the PAMAM-calix-dendrimers of the first generation, the compounds G1-cone, G1-paco, and G1-1,3-alt, were obtained (Scheme 1) with quantitative yields (96–99%).
Thus, an efficient method for the synthesis of the first generation PAMAM-calix-dendrimers dendrimers G1 and their precursors (half-generation) G0.5 has been developed. The structure and composition of the obtained compounds G0.5 and G1 were confirmed by 1H, 13C NMR, IR spectroscopy, mass spectrometry, and elemental analysis.
NMR spectroscopy is a classic technique for establishing the structure of macromolecules as well as for evaluating the purity of these compounds [39]. The PAMAM dendrimers have a low number of signals in the 1H and 13C NMR due to their highly symmetrical and regular structure. It is known that various conformations of thiacalix[4]arene differ in chemical shifts of the proton signals due to the influence of the macrocyclic platform [39,40,41]. That allows to unequivocally determine the conformation of the macrocycle by 1H NMR spectroscopy. The strongest differences were observed in the chemical shifts of the proton signals of amidomethylene and oxymethylene fragments closest to the p-tert-butylthiacalix[4]arene platform. In the case of the amphiphilic-like compound G1-cone with asymmetric structure (Figure 1), these signals were located at 3.33 and 4.89 ppm, respectively. In the case of symmetric dendrimer G1-1,3-alt, these groups were shielded by neighboring aromatic rings of the macrocycle, and their signals appeared at 3.25 and 4.15 ppm. Smaller differences were observed in the chemical shifts of the proton signals of the tert-butyl and aryl fragments. Compared to G1-1,3-alt, chemical shifts of the aryl protons in the G1-cone drifted upfield (7.45 ppm for G1-cone against 7.59 ppm for G1-1,3-alt) due to the shielding effect of neighboring aryl fragments on the aryl protons of the macrocycle ring. Similarly, the peaks of the tert-butyl groups of the G1-cone were also shifted upfield (1.14 ppm) compared to corresponding peaks for the G1-1,3-alt (1.28 ppm).
In the case of the asymmetric compound G1-paco, symmetry of the macrocyclic core was disturbed, and this complicated the spectra. The peaks of the tert-butyl fragment appeared as two singlets at 1.09 and 1.35 ppm with a ratio of 1:1, peaks of protons of aromatic and oxymethylene groups formed two singlets and AB system.
It should be noted that many chemical shifts of branching proton signals coincided in comparison to the 1H NMR spectra of three compounds G1-cone, G1-paco, and G1-1,3-alt. This occurs even in the NMR spectrum of the compound G1-paco, although the NMR spectra of the thiacalix[4]arene derivatives in partial cone conformation are usually complicated due to the disturbance of the symmetry of the macrocyclic platform. Thus, in all three stereoisomers, the proton signals of the methylene groups of branching appeared in the same areas (2.36, 2.46, 2.65–2.80, and 3.24 ppm). This is probably due to the decreasing influence of the macrocycle conformation resulting from the distancing of mentioned groups from the macrocyclic platform. Besides, the obtained NMR data indicates the purity of the synthesized first-generation PAMAM-calix-dendrimers and the absence of defects in their structure.
The obtained compounds G0.5 and G1 were also characterized by mass spectrometry. For example, ESI high-resolution mass spectra of the compounds G1 showed peaks corresponding to molecular ions with three, four, five and six protons ([M + 3H]3+, [M + 4H]4+, [M + 5H]5+, and [M + 6H]6+).
Thus, the novel first generation of PAMAM-calix-dendrimers was designed and synthesized with high yields. The structure of the obtained compounds was proven by modern physical methods.
This entry is adapted from the peer-reviewed paper 10.3390/ijms222111901
This entry is offline, you can click here to edit this entry!