Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Public, Environmental & Occupational Health
|
Biochemistry & Molecular Biology
|
Orthopedics
氟在自然界中广泛分布,具有多种生理功能。虽然它通常被认为是人类必不可少的微量元素,但这一观点并不普遍。此外,慢性氟中毒,主要以骨骼氟化物为特征,可由长期过量的氟化物消费引起。环境中氟化物浓度高和饮用水是主要原因,骨骼氟化物患者主要表现为骨硬化、骨质疏松症、骨质疏松症、骨质疏松症和关节软骨退行性变化。骨骼氟化物的病因已经确定,但具体的发病机制没有定论。目前,活跃的骨骼生成和加速骨周转被认为是骨骼氟化进展的关键过程。近年来,研究人员在信号通路领域进行了广泛的研究(Wnt/β-卡特宁, 诺奇、PI3K/Akt/mTOR、刺猪、甲状旁腺激素和胰岛素信号通路)、应激通路(氧化应激和内质视网膜应激通路)、表观遗传学(DNA甲基化和非编码RNA),及其参与骨骼氟化发病机制的相互调节。
- skeletal fluorosis
- fluoride
- endemic disease
- signaling pathways
- epigenetics
- endoplasmic reticulum stress
- oxidative stress
1. 简介
氟是最常见的卤素之一,通常作为化合物存在于环境中。氟或氟化物主要通过饮用水、食物和空气进入人体。一旦被吸收到血液中,它很容易以离子的形式在全身传播。超过90%的吸收氟化物分布在骨组织中,正常人体中的氟总量约为2.6克[1].氟化物对人类福祉有双重影响。人体的正常生长发育需要微量的氟化物[2],而高氟化物的摄入量可能会对组织、器官和系统造成损害。牙氟病和骨骼氟化是氟化的典型表现。长期饮用高氟水[2]和砖茶[3][4],或因使用高氟煤作为燃料而进入人体的受污染空气和食物[5][6]可导致氟中毒。不幸的是,对于严重威胁人类健康的骨骼氟化物患者,没有明确有效的治疗方法。尽管近年来发病率不断下降,但全球40多个国家的氟中毒,特别是骨骼氟病,仍然是一个严重的公共卫生问题。因此,认识到骨骼氟化进展的机制至关重要。
氟具有很强的骨骼亲和力,往往积聚在骨组织中,导致骨骼氟化。过量的氟化物会对人体的骨细胞、骨质疏松物、软骨组织和骨矿化造成一系列损害[7].氟在骨骼生成和吸收中具有双向特征:它不仅能通过增强骨质活性而引起骨硬化,还能通过促进骨吸血导致骨质疏松症[8].迄今为止,尚未充分探讨骨骼氟化物的发病机制。多年来,研究人员一直关注氟影响骨周转过程的各种细胞调节机制[9].积极的骨生成和加速骨周转已被证明是骨骼氟化进展的关键过程和骨源病变多样性的病理基础[9][10].氟可以诱发骨细胞和骨质疏松症的分化和凋亡,主要通过破坏骨周转的动态平衡,导致骨骼损伤,并最终导致骨硬化、骨质疏松、腹膜软组织骨质疏松、骨质疏松症,以及骨骼氟化患者的关节和软骨退行性变化。
近年来,研究人员在了解信号通路、氧化应激、内质视网膜应激、DNA甲基化和非编码RNA的调控方面取得了重大进展。通过分析涉及骨骼氟病发机制的相关信号通路、应激通路、表观遗传学和相互作用网络的若干研究,本审查旨在加深对氟病发病机制的理解,并有望提出更有针对性的预防和治疗策略。
2. 骨骼氟化物信号通路机制
2.1. Wnt/β-卡特宁信号通路对骨骼氟病的影响
Wnt 是一种参与各种生物过程的细胞因子[11],规范的Wnt/β-catenin信号通路在调节骨细胞分化、骨原基质形成和骨平衡方面起着至关重要的作用[12][13][14][15][16].但是,在没有 Wnt 配体的情况下,路径的潜在转录机制无法激活[17].当 Wnt 配体 (Wnt1, 1) 时, 糖原合成酶激酶 - 3β (GSK-3+) 的β- 卡特宁抑制作用受到阻碍 Wnt2、Wnt3a 和 Wnt10a 作用于细胞表面受体,这些受体具有与脂蛋白相关蛋白质 5/6 (LRP5/6) 的模糊和低密度脂蛋白受体作用,导致β-卡特宁的磷酸化和降解降低。因此,β-卡特宁能够积累到足够高的水平进入细胞核并与T细胞因子/淋巴增强因子(TCF/LEF)相互作用,从而调节目标基因的表达[17].
Sun等人证实,氟化物显著增加了骨细胞活性,这从他们研究中观察到的增强性骨生成中得到了证明[18].氟化物增加了大鼠骨细胞中Wnt3a和β-卡特宁的表达:此外,血清骨碱性磷酸酶(BALP)水平往往随着氟化物染色剂量的增加而增加[19],建议氟化物可能参与骨骼形成过程,刺激Wnt3a向上调节BALP表达。最近的一项研究还发现,氟化物可诱导Wnt/β-卡特宁信号通路的异常激活,导致小鼠癌骨的形成和Wnt3a和磷-Gsk-3的蛋白质表达增加,以及其下游目标基因Runt相关转录因子2(Runx2)。然而,必须指出,抑制β-卡特宁可以抑制氟化物引起的Runx2蛋白质表达和由此产生的骨病理学[15],这表明β-卡特宁可能是氟化物诱发异常骨生成的关键分子。血清硬化蛋白 (SOST) 和迪克科普夫相关蛋白质 1 (Dkk-1) 是 Wnt/β-卡特宁信号的抑制剂,和王[20]刘[21],和曾等人。[22]结果表明,长期或高浓度接触氟化物可降低SOST和Dkk-1浓度,从而诱导Wnt/β-卡特宁信号的激活,最终导致骨细胞的进展和分化。图1显示了Wnt/β-卡特宁信号通路与骨骼氟化发病机制之间的关联。
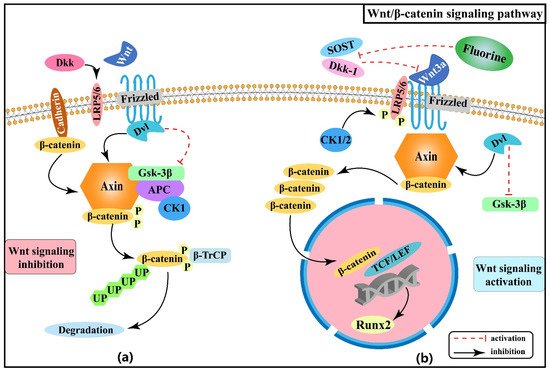
图1。骨骼氟化物发病机制中的Wnt/β-卡特宁信号通路。(a) Wnt 信号被抑制:(b) 启动 Wnt 信号。
在这里,我们回顾了Wnt/β-卡特宁信号通路在骨骼氟中毒中的作用机制,很明显,这种信号可以调解氟化物增强的骨源效应。此外,Wnt/β-卡特宁信号通路与控制骨细胞和软骨细胞增殖和分化的许多信号通路相连,如PIK/Akt和刺猪(Hh)信号,随后对探索其交互调节的研究对于理解骨骼氟化病的发病机制更有价值。
2.2. Effect of Notch Signaling Pathway on Skeletal Fluorosis
Notch signaling pathway primarily mediates intercellular interactions and plays an integral part in determining cell fate and function together with the regulation of skeletal homeostasis [23]. The interactions of Notch receptors (Notch-1, 2, 3, and 4) with ligands (Jagged-1, Jagged-2, and Delta-like-1, 3, and 4) lead to a series of proteolytic cleavage and release of the Notch intracellular structural domain (NICD) into the cytoplasm [24][25]; NICD translocates to the nucleus and forms a complex with Epstein-Barr virus latency C promoter binding factor 1 (CBF-1)/repressor of hairless/Lag1 (CSL), which is known as recombination signal binding protein-Jκ (RBP-J) in mice, and Mastermind-like (Maml) to activate transcription of target genes (Hes-1, Hes-5, Hey-2, and Hey L) [26][27][28]. Notch signaling pathway regulates the development of osteoblast and osteoclast lineages and thus has a significant impact on skeletal development [29].
Notch signaling can inhibit osteoblast differentiation [30][31], thereby maintaining bone marrow mesenchymal progenitors [27]. An animal experiment found that excessive fluoride exposure decreased the protein as well as mRNA expression levels of Notch-3 and Jagged-1 in rats, especially in osteoblasts [32], suggesting that fluoride can inhibit the Notch signaling pathway, thus promoting osteoblasts proliferation and differentiation, disturbed dynamic homeostasis of bone tissue, and the pathological manifestation of osteosclerosis. Another study [33] showed that RBP-J protein expression was increased and Hes-5 protein expression was decreased in bone tissue of rats dyed with fluoride, indicating that fluoride may promote osteoblast differentiation by promoting the expression of the transcriptional repressor RBP-J and enhancing the inhibitory effect on the downstream target gene Hes-5. Prior studies have suggested that the Notch signaling pathway mediates the enhanced osteogenic effects of fluorosis. Figure 2 shows the association between Wnt/Notch signaling pathway and the pathogenesis of skeletal fluorosis.
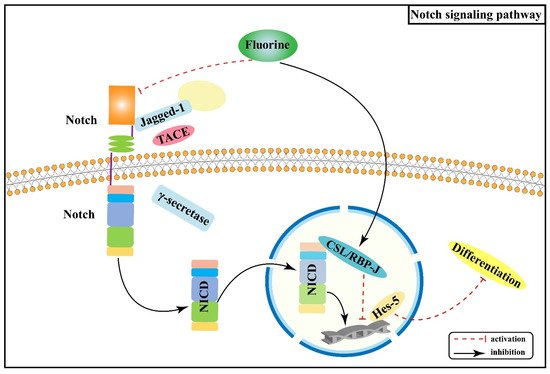
Figure 2. Notch signaling pathway in the pathogenesis of skeletal fluorosis.
Notch signaling pathway displays a significant role in cell proliferation, differentiation, and apoptosis. Nonetheless, there are relatively few investigations on the relationship between Notch signaling pathway and skeletal fluorosis, mainly focusing on the mechanisms that regulate the proliferation and differentiation of osteoblasts. An in-depth investigation of the role of Notch signaling pathway in regulating other bone tissues and cells and its synergistic or antagonistic effects with other signaling pathways is of great significance for the pathogenesis, prevention, and treatment of skeletal fluorosis.
2.3. Effect of PI3K/Akt/mTOR Signaling Pathway on Skeletal Fluorosis
mTOR is a serine/threonine-protein kinase that can regulate a variety of biological processes. PI3K/Akt/mTOR signaling pathway is involved in the proliferation and differentiation of osteoblasts, osteoclasts, and chondrocytes [34][35][36]. Experiments in rats with chronic fluorosis have shown that the PI3K/Akt signaling may cause excessive proliferation and differentiation of osteoblasts and accelerate bone turnover, resulting in osteosclerotic skeletal fluorosis [37]. It was found that high-dose fluorine can inhibit the expression of mTOR protein in cartilage tissue, promote the expression of autophagy signature proteins, such as Beclin-1 and cytoplasm-associated protein light chain 3 (LC3), and enhance the apoptosis involved in the process of fluorine-induced cartilage damage [38]. After mTOR is activated, it phosphorylates the downstream target proteins ribosomal protein S6 kinase β-1 (S6K1) and eukaryotic translation initiation factor 4E binding protein 1 (4EBP1) to promote gene transcription and protein translation [39]. Fluorine can down-regulate PI3K/Akt/mTOR signaling and inhibit the phosphorylation of its downstream factors S6K1 and 4EBP1, thus promoting chondrocyte autophagy and inhibiting chondrocyte proliferation and differentiation [40]. The association between PI3K/Akt/mTOR signaling pathway and the pathogenesis of skeletal fluorosis is shown in Figure 3.
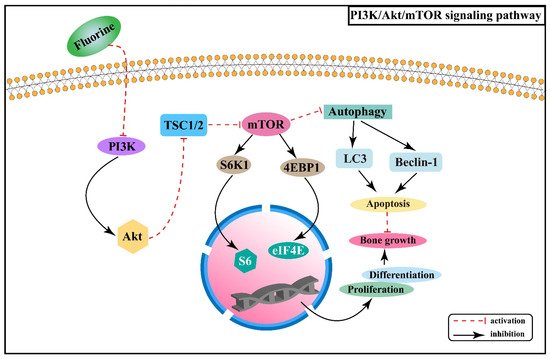
Figure 3. PI3K/Akt/mTOR signaling pathway in the pathogenesis of skeletal fluorosis.
Skeletal fluorosis is characterized by a disrupted dynamic balance between bone resorption and formation and is intimately linked to regulating PI3K/Akt/mTOR signaling pathway, with a critical role in osteoblast proliferation and differentiation as well as chondrocyte autophagy. Understanding more about the changes of PI3K/Akt/mTOR signaling pathway and related signaling molecules in skeletal fluorosis, as well as its interaction with other signaling pathways, will promote a profound understanding of the pathogenesis of endemic fluorosis and provide a basis for the prevention and treatment of skeletal fluorosis.
2.4. Effect of Hedgehog Signaling Pathway on Skeletal Fluorosis
Hh signaling pathway consists of Hh protein, protein receptors Ptched (Ptch) and Smoothened (Smo), and the 5-zinc finger transcription factor Glis (Gli1, Gli2, and Gli3) [41]. There are three Hh homolog genes in mammals: Sonic Hedgehog (Shh), Indian Hedgehog (Ihh), and Desert Hedgehog (Dhh), which encode Shh, Ihh, and Dhh proteins, respectively. In the absence of Hh ligands, Ptch inhibits the activity of Smo, which further inhibits the expression of downstream genes in the nucleus [42]. When the Hh ligand binds to Ptch, its inhibitory effect on Smo is released, and the expression of target genes increases [42]. Research results have shown that removing Smo decreased the proliferation rate of chondrocytes, while upregulation could increase the rate [43].
Studies have revealed that fluoride stimulation increases the expression of Ihh in rat osteoblasts, and this signal, mediated by Smo, induces the expression of Runx2 after activating Gli2, which in turn promotes osteoblast proliferation and contributes to osteosclerosis [44]. Moreover, fluorine stimulation can cause chondrocytes to produce oxygen radicals, thus causing abnormal differentiation [45][46]. Shh is stimulated and released when oxidative stress occurs, synergizing with oxidative stress to promote the B cell lymphoma/leukemia-2 (Bcl-2) anti-apoptotic protein [47]. Animal experiments demonstrated that with the expression of Shh, Smo, bone morphogenetic protein-2 (BMP-2) and B cell lymphoma/leukemia gene associated × (Bax) protein gradually increased in rat cartilage tissues with increasing fluoride staining concentration, while Bcl-2 protein expression gradually decreased [48]. Figure 4 shows the association between Hh signaling pathway and the pathogenesis of skeletal fluorosis.
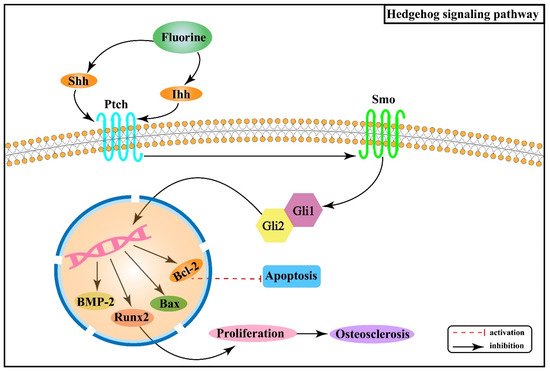
Figure 4. Hedgehog signaling pathway in the pathogenesis of skeletal fluorosis.
The above studies suggest that the Hh signaling pathway is closely associated with oxidative stress and apoptosis and regulates chondrocyte ossification and apoptosis in skeletal fluorosis. In addition, the available data reveal that Hh signaling is also related to Wnt/β-catenin, Notch, Bmp, and other signaling pathways, and its specific regulatory mechanisms in the pathogenesis of skeletal fluorosis that damages cartilage tissue remain to be investigated in depth.
2.5. Effect of Hormones and Their Receptor Signaling Pathways on Skeletal Fluorosis
Over the years, systematic studies of the mechanisms of fluorosis in animals and humans have revealed that many calcium-regulating hormones and cytokines are also involved in mediating abnormal osteogenic and osteolytic bone metabolism in skeletal fluorosis, such as parathyroid hormone (PTH) and transforming growth factor β (TGF-β). PTH is a polypeptide hormone responsible for regulating calcium homeostasis in the body and exerts an active role in regulating bone turnover. Its effects on cells are mediated by the G protein-coupled receptor (PTH receptor, PTH1R) expressed by target cells, including osteoblasts, osteoblast precursor cells, and osteocytes, but excluding osteoclasts [49][50]. Therefore, PTH acts indirectly on osteoclasts by binding to PTH1R on the surface of the osteoblast cell line and secreting osteoclast differentiation factors. Additionally, a study found that serum PTH levels gradually increased with increasing fluoride exposure [51]. PTH can regulate fluoride action through its effects on the expression of SOST and receptor activator of nuclear factor-κB ligand (RANKL) in osteocytes. By up-regulating RANKL and inhibiting SOST, PTH enhances the effectiveness of low-dose of fluoride in bone turnover promotion while having opposite effects on SOST and RANKL in the cases of high fluoride doses [52]. Figure 5 shows the association between PTH signaling pathway and the pathogenesis of skeletal fluorosis.
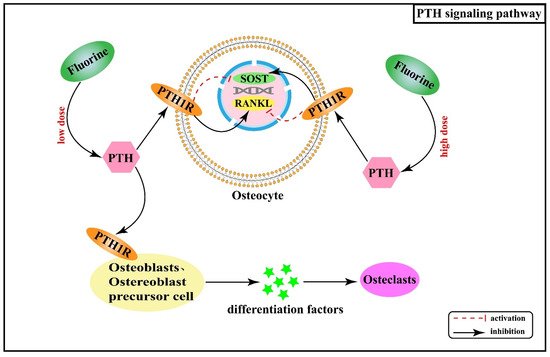
Figure 5. PTH signaling pathway in the pathogenesis of skeletal fluorosis.
In addition, PTH, as one of the important hormones regulating calcium and phosphorus metabolism in the body, exerts a bidirectional regulatory effect on bone metabolism. High doses of PTH promote bone resorption, while at low doses, it stimulates bone formation. Furthermore, animal and clinical studies have revealed that when PTH is given intermittently subcutaneously, it stimulates bone formation, increases bone density in long bones and vertebrae, and improves the bone quality [53][54]. It follows that the use of PTH in the treatment of osteoporosis has been approved by the US Food and Drug Administration and other organizations [54].
Insulin is a multifunctional protein hormone that acts by binding to the insulin receptor (IR) in most tissues. IR and intracellular signaling pathways are the main components of the insulin signaling pathway [55]. Fulzele et al. [56] found that IR exists in osteoblasts, and insulin promotes bone formation by inhibiting Twist2 (Runx2 inhibitor) after interacting with cell surface receptors. Studies have pointed out that fluoride can stimulate IR expression and osteoblast function in vitro and affect insulin secretion, activity, and sensitivity [57][58]. Furthermore, insulin state in turn interferes with bone formation and absorption [57][58]. Another study showed that insulin plays a vital role by mediating IR signals, and insulin-like growth factor-1 (IGF-1) may play a role in bone turnover induced by excessive fluoride by regulating IR or its downstream [59]. All these studies suggest that insulin exerts an essential role in fluoride-induced bone pathogenesis.
2.6. Interactive Regulatory Networks among Signaling Pathways Involved in Skeletal Fluorosis
In the pathogenetic progression of skeletal fluorosis, both local signaling pathways and hormones are involved. Activation or inhibition of signaling is not accomplished linearly within the single signaling pathway mentioned above alone, but rather the signaling pathways interact to form an interactive regulatory network that acts to control biological processes in a synergistic or antagonistic manner [60]. Figure 6 illustrates the interactive regulatory networks formed among Wnt/β-catenin, Notch, PI3K/Akt/mTOR, Hh, PTH, and insulin signaling pathways involved in skeletal fluorosis.
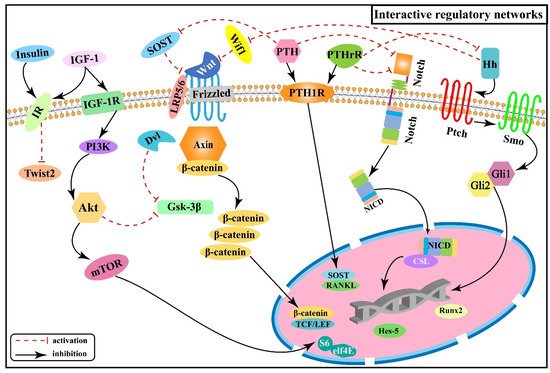
Figure 6. Interactive regulatory networks among Wnt/β-catenin, Notch, PI3K/Akt/mTOR, Hh, PTH, and insulin signaling pathways involved in skeletal fluorosis.
PTH can bind to the same receptor (PTH1P) as the parathyroid hormone-related peptide (PTHrP) synthesized by osteoblasts and mediates bone formation and accelerated bone turnover through multiple pathways. And SOST expressed by osteoblasts is a key negative regulator of bone formation [61][62] and acts as an upstream inhibitor of the Wnt/β-catenin signaling pathway by binding to Wnt and blocking its interaction with the cell surface receptors Frizzled or LRP5/6 to inhibit the activation of β-catenin downstream in the Wnt signaling pathway. Whereas PTH causes transcriptional suppression of the osteocyte marker gene SOST and downregulates SOST protein expression [63], thereby activating the Wnt/β-catenin signaling pathway. Experiments revealed that the expression of Ihh in rat growth plate chondrocytes decreased with increasing fluorine concentration, while the expression of PTHrP showed an increasing trend, suggesting that fluorine reduces the expression of Ihh by up-regulating PTHrP, inhibiting the Ihh/PTHrP negative feedback loop to affect chondrocyte proliferation and differentiation, and inhibiting the normal process of osteogenesis within cartilage [64]. In addition, it was shown that PTH can down-regulate the expression of the Notch signaling pathway, and this inhibitory effect may be achieved by down-regulating intracellular cAMP/PKA signaling, reducing the expression of receptor Notch-1 and ligand Jagged-1 of the Notch signaling pathway, which in turn affects the expression of Runx2 [65]. The researchers found positive regulation among PTHrP, NICD, and Jagged-1 proteins after transfecting epiphyseal stem cells with PTHrP over-expression and lentiviral interference vectors, suggesting that PTHrP may act through influencing the Notch signaling pathway [66]. The above information indicates that the Wnt/β-catenin, Hh, and Notch signaling pathways are associated with the PTH signaling pathway and act downstream of the PTH signaling.
Both Wnt/β-catenin and Ihh signaling pathways control the proliferation and differentiation of osteoblasts and chondrocytes at multiple stages [12][13][14][15][44][45][46]. One study tested the genetic relationship between Wnt and Hh signaling by generating double mutant mice with results revealing that Wnt/β-catenin signaling acts downstream of Hh signaling in enhancing bone formation [67], in agreement with another study [68]. Subsequently, the expression of genes associated with different stages of osteoblast differentiation was also examined, showing that β-catenin is required downstream of Ihh in promoting osteoblast maturation [67]. It has been demonstrated that Wif1 (Wnt inhibitory factor 1) exerts biological effects by mediating and regulating Shh/Wnt/β-catenin signaling [69], as well as Wif1 can effectively block the activation of the canonical Wnt signaling pathway in chondrocytes by binding to Wnt ligands (Wnt3a, etc.) [69][70][71], speculating Hh signaling may exert inhibitory effects on downstream Wnt/β-catenin signaling through Wif1 in the pathogenesis of skeletal fluorosis. At the same time, the Wnt/β-catenin signaling is also interactively regulated with the PI3K/Akt signaling. It was shown that PI3K/Akt negatively regulates Gsk-3β activity, thereby inhibiting the phosphorylation of β-catenin [72], which suggests a role for Wnt/β-catenin signaling acting downstream of PI3K/Akt. To determine this role, the researchers further analyzed the expression of Wnt-regulated genes, such as Dkk1 and Sfrp1 (secreted frizzled-related protein 1), showing that PI3K signaling activates these genes by peptide-mediated α5β1 integrin priming in mesenchymal skeletal cells [73]. Moreover, PI3K/Akt signaling is also regulated by insulin-related signaling. IGF-1 is a principal growth-promoting signal for vertebrate skeletal development [74] and as a specific ligand can activate the PI3K/Akt signaling pathway by binding and phosphorylating the membrane IGF-1 receptor (IGF-1R), leading to osteoblast differentiation and proliferation [75].
At present, studies on the relevant signaling pathways involved in skeletal fluorosis are still predominantly single lineage studies. Nevertheless, active osteogenesis and accelerated bone turnover as crucial processes in the progression of skeletal fluorosis are regulated by sophisticated networks of multiple signaling pathways. The profound exploration of the interactive regulatory mechanisms among signaling pathways in skeletal fluorosis will facilitate the development of specific targeted therapeutic measures.
3. Mechanism of Stress Pathways in Skeletal Fluorosis
3.1. Effect of Endoplasmic Reticulum Stress on Skeletal Fluorosis
The endoplasmic reticulum (ER) is essential for protein synthesis and secretion in eukaryotic cells [76]. When cells are subjected to certain external stimuli, the ER generates a series of regulatory mechanisms, generating endoplasmic reticulum stress (ERS) [77]. At the same time, ERS can protect cells from damage by stimulating the unfolded protein response (UPR) [78] or initiating the apoptotic program to ensure the survival of the organism [79][80]. Proteomic studies have revealed that the expression of immunoglobulin heavy chain binding protein (BiP), also known as glucose-regulated protein 78 (GRP78), protein disulfide isomerase (PDI), proteasome 26S ATPase, and thioredoxin (Trx) is up-regulated in fluoride-stained osteoblasts, and these proteins play key roles in protein folding of ER [81]. The results provide clues that ERS and UPR may be involved in the pathogenesis of skeletal fluorosis. URP is mediated by Bip and three response sensing proteins, including protein kinase-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6) [82].
PERK is a serine/threonine-protein kinase that can be activated by accumulating misfolded or unfolded proteins on the ER [83]. It can reduce the newly synthesized proteins of the ER by activating eukaryotic initiation factor 2a (eIF2a) and activating transcription factor 4 (ATF4) pathways, thereby reducing ER load, and even inducing C/EBP homologous protein (CHOP) protein synthesis to stimulate the apoptotic program [84][85][86][87]. In addition, PERK can also directly activate nuclear factor erythroid 2-related factor 2 (Nrf2) to antagonize apoptosis to protect cell survival [88]. A study has shown that fluoride exposure activates the PERK signaling pathway, leading to activation of ATF4 and Nrf2 and up-regulating the expression of genes related to bone turnover in osteoblasts [89]. Meanwhile, the study also showed that ATF6 and IRE1 signaling factors mediated a less pronounced effect of ERS, suggesting that fluoride exposure mediates osteoblast damage mainly through the PERK signaling pathway [89].
To further clarify the role of the PERK signaling pathway, changes in osteogenic and osteolytic gene expression were studied in osteoblastic cell lines before and after PERK gene interference, and the results showed that fluoride stimulated the protein expression of PERK, Nrf2, osteoprotegerin (OPG), and Runx2 in PERK siRNA-transfected cells to a certain extent [90]. The above findings suggest the vital role of the PERK/Nrf2 pathway in the activation mechanism of fluorine-induced osteoblast and osteoclast. Nevertheless, studies on the IRE1 pathway and ATF6 pathway concerning skeletal fluorosis are still scarce and need to be further explored to provide new ideas and theoretical basis for preventing and treating skeletal fluorosis.
3.2. Effect of Oxidative Stress on Skeletal Fluorosis
Oxidative stress is considered to be an essential mechanism in the pathogenesis of fluorosis. Intake of large amounts of fluoride can cause an imbalance between antioxidant defense mechanisms and free radical levels, leaving cells in a state of oxidative stress [91]. In fluorosis, oxidative stress increases reactive oxygen species (ROS) and is accompanied by reduced antioxidant enzyme activity and increased lipid peroxides [92][93]. Nevertheless, excessive ROS can disrupt the dynamic balance between bone formation of osteoblasts and bone resorption of osteoclasts, leading to the development of skeletal fluorosis [94]. A study has shown that osteoblasts treated with low fluorine concentrations are in a low-level oxidative stress state and maintain redox homeostasis by activating the nuclear factor E2-related factor 2-antioxidant responsive element (Nrf2-ARE) signaling pathway, which prevents cells from apoptosis or death [95]. In contrast, when treated with high concentrations (4.00 mmol/L) of fluoride, the intracellular antioxidant system and signaling pathways were disrupted or disintegrated, leaving the cells in a decompensated state, and undergoing severe oxidative damage, even inducing apoptosis in osteoblasts [95]. Another study noted that intracellular ROS levels increased significantly after sodium fluoride treatment, and osteoblasts presented apoptotic morphological changes such as chromatin condensation and DNA fragmentation [96].
In addition, fluoride can likewise participate in the regulation of osteoclast proliferation through the oxidative stress pathway. Calcineurin (CaN) is a serine/threonine phosphatase dependent on calcium (Ca) and calmodulin (CaM), which is involved in the regulation of osteoclast differentiation and proliferation [97]. Animal experiments have demonstrated that excessive fluoride exposure led to increased CaN mRNA and protein expression levels and serum CaN activity in rat bone tissue, but its upstream regulators Ca and CaM had no significant differences between the control and fluoride-infected groups [98][99]. However, malondialdehyde (MDA) levels of the fluoride-infected group were significantly higher than the control group, and oxidative stress levels were also positively correlated with CaN activity [99][100], suggesting excess fluoride may stimulate elevated CaN activity in the organism through the oxidative stress pathway, further leading to increased osteoclast production in rat bone tissue.
The above studies have illustrated that fluoride can induce osteoblast apoptosis and osteoclast proliferation through the oxidative stress pathway, providing evidence for the pathogenesis of skeletal fluorosis. However, the molecular mechanisms of fluoride effects on osteoclasts are still relatively poorly investigated, and how fluoride affects osteoclast proliferation and differentiation through the oxidative stress pathway has not been comprehensively clarified, and further in-depth studies are necessary. Besides, since both endoplasmic reticulum stress and oxidative stress are associated with the pathogenesis of skeletal fluorosis, further studies are needed to explore the interrelationship between them to provide new insights into the pathogenesis of skeletal fluorosis.
4. Mechanism of Epigenetics in Skeletal Fluorosis
4.1. Effect of DNA Methylation on Skeletal Fluorosis
DNA methylation is one of the earliest and most common epigenetic modifications, and it plays an important role in the pathogenesis of skeletal fluorosis. P16 protein is a key factor of cell cycle regulation in the G1/S phase, competitively inhibits the cell progression from G1 phase to S phase and blocks abnormal activation of osteoblasts [101][102]. In fluorosis model experiments, promoter hypermethylation of the p16 gene inhibited its mRNA transcription and protein expression, while decreased p16 protein expression reduced its G1/S phase blocking effect, potentially providing an important molecular mechanism for the altered proliferative capacity and cell cycle distribution of osteoblasts caused by fluorine [103]. In addition, research has revealed that fluoride causes promoter DNA hypermethylation of the BMP1, METAP2, MMP11, and BACH1 genes, then RNA expression levels are down-regulated, thereby promoting skeletal fluorosis [104].
An animal experiment has shown that estrogen receptor α (ERα) mRNA expression is enhanced when the promoter methylation level of the ERα gene is inhibited in osteoblasts, thereby promoting osteoblast proliferation and differentiation [105]. In addition, the promoter methylation level of the ERα gene is negatively correlated with urinary fluoride concentration, suggesting that the pathological bone changes induced by fluoride exposure in men were related to the promoter hypomethylation of the ERα gene [106]. In conclusion, due to the diversity of fluoride regulation of bone damage and cellular DNA methylation, further works on the epigenetics of cellular regulation-related genes in fluorosis are needed.
4.2. Effect of Non-Coding RNAs on Skeletal Fluorosis
Following extensive research on the molecular mechanisms of abnormal bone metabolism in skeletal fluorosis at the molecular biology level, the role of microRNAs (miRNAs) is gradually being discovered. miRNAs as non-coding RNAs are post-transcription regulators of gene expression. miRNAs negatively regulate the expression of their target mRNAs and can inhibit the expression of mRNAs [107]. Fluoride exposure in human osteosarcoma cells can affect the expression of genes related to bone metabolism through the miRNA pathway [108]. Cyclin D1 is a protein that regulates the cell cycle by facilitating the cell transition from G1 phase to S phase and accelerating cell proliferation [109]. Down-regulation of mir-486-3p can affect the expression of cyclin D1 and further regulate osteoblast proliferation and activation [110]. Studies have shown that fluoride exposure induces the down-regulation of miR-4755-5p and Let-7c-5p, promoting fluoride-induced osteoblast proliferation and activation by regulating cyclin D1 expression [111][112].
It was pointed out that miR-29a promotes the differentiation process of osteoblasts by targeting and inhibiting the expression of Dkk-1 [113]. The specific mechanism is that the canonical Wnt signaling pathway can induce transcription of miR-29a, which can enhance the Wnt signaling pathway by downregulating Dkk-1 (an antagonist of Wnt signaling), thereby regulating the osteoblast differentiation process [113][114]. Another study also showed that the expression level of miR-27 was positively correlated with that of β-catenin, and it could activate Wnt signaling through the accumulation of β-catenin protein, thereby promoting osteoblast differentiation [115]. This suggests that miR-27 could be a possible target for the development of drugs against skeletal fluorosis. In conclusion, the regulatory networks of non-coding RNAs in fluorosis are complex, and further investigations are needed to elucidate their regulatory mechanisms on specific target mRNAs. Meanwhile, exploring the interactions between miRNAs and other signaling pathways may provide new insights into the therapeutic strategies for skeletal fluorosis.
5. Conclusions
In summary, skeletal fluorosis is a chronic and progressive endemic disease with severe risks to human health. Exposure to a specific duration or dose of fluoride can alter osteoblasts, osteoclasts, and chondrocytes’ function, differentiation, and proliferation and lead to skeletal fluorosis by disrupting the balance between bone formation and resorption. Signaling pathways, stress pathways, epigenetics, and interactive regulatory mechanisms play crucial roles in the pathogenesis above. With the development of multi-level and multi-faceted studies in recent years, considerable progress has been made at the molecular biological level in understanding the pathogenesis of skeletal fluorosis. Unfortunately, although a great deal of research has been carried out, there are no clear and effective treatments for patients with skeletal fluorosis.
The treatment of skeletal fluorosis in traditional Chinese medicine (TCM) focuses on reducing fluoride concentrations in the body and improving autogenous regulatory mechanisms, while western medicine aims to reduce the toxicity of fluoride to the body and repair the damage through specific interventions [116]. Studies have suggested that chemical elements such as calcium and magnesium can antagonize fluoride toxicity by binding fluoride in the intestine to form low-solubility complexes for excretion, thereby reducing fluoride absorption [117]. Antioxidants have also been used to treat skeletal fluorosis, and concomitant intake of calcium, vitamin C, and vitamin D can antagonize fluoride toxicity [118]. Additionally, some components of Chinese herbal medicine have antioxidant effects and can also effectively antagonize the oxidative stress caused by fluorosis, thereby reducing the degree of fluorosis [94][119]. Considering the combination of TCM and Western medical treatment ideas for skeletal fluorosis may yield better effects. At the same time, it is more important to continue in-depth research on the complex pathogenesis of skeletal fluorosis and explore new targeted therapies at the molecular level to treat skeletal fluorosis.
Since there is no specific effective treatment to cure skeletal fluorosis completely, prevention and control of skeletal fluorosis by using safe drinking water and reducing the use of high fluoride coal burning is currently considered the ideal approach [2]. Particularly, clarifying the pathogenesis of skeletal fluorosis and exploring effective targets against fluorine toxicity are of great importance to scientifically prevent and control the occurrence, development, and prevalence of skeletal fluorosis and reduce the risk to human health.
This entry is adapted from the peer-reviewed paper 10.3390/ijms222111932
References
- Zhao, Y.Y. The Progress about the influence of Fluorine on Bone. Med. Recapitul. 2006, 12, 1092–1094.
- Srivastava, S.; Flora, S.J.S. Fluoride in Drinking Water and Skeletal Fluorosis: A Review of the Global Impact. Curr. Environ. Health Rep. 2020, 7, 140–146.
- Fan, Z.P.; Gao, Y.H.; Wang, W.; Gong, H.Q.; Guo, M.; Zhao, S.C.; Liu, X.H.; Yu, B.; Sun, D.J. Prevalence of Brick Tea-Type Fluorosis in the Tibet Autonomous Region. J. Epidemiol. 2016, 26, 57–63.
- Izuora, K.; Twombly, J.G.; Whitford, G.M.; Demertzis, J.; Pacifici, R.; Whyte, M.P. Skeletal fluorosis from brewed tea. J. Clin. Endocrinol. Metab. 2011, 96, 2318–2324.
- Ando, M.; Tadano, M.; Asanuma, S.; Tamura, K.; Matsushima, S.; Watanabe, T.; Kondo, T.; Sakurai, S.; Ji, R.D.; Liang, C.K.; et al. Health effects of indoor fluoride pollution from coal burning in China. Environ. Health Perspect. 1998, 106, 239–244.
- Xu, Y.Y.; Huang, H.; Zeng, Q.B.; Yu, C.; Yao, M.L.; Hong, F.; Luo, P.; Pan, X.L.; Zhang, A.H. The effect of elemental content on the risk of dental fluorosis and the exposure of the environment and population to fluoride produced by coal-burning. Environ. Toxicol. Pharmacol. 2017, 56, 329–339.
- Mou, W.P.; Yan, H.; Zhang, L.H. Progress in molecular mechanism of skeletal fluorosis. Chin. Foreign Med. Res. 2011, 9, 158–160.
- Noël, C.; Gosselin, B.; Dracon, M.; Pagniez, D.; Lemaguer, D.; Lemaître, L.; Dhondt, J.L.; Lelièvre, G.; Tacquet, A. Risk of bone disease as a result of fluoride intake in chronic renal insufficiency. Nephrologie 1985, 6, 181–185.
- Wei, W.; Pang, S.J.; Sun, D.J. The pathogenesis of endemic fluorosis: Research progress in the last 5 years. J. Cell Mol. Med. 2019, 23, 2333–2342.
- Boivin, G.; Chavassieux, P.; Chapuy, M.C.; Baud, C.A.; Meunier, P.J. Skeletal fluorosis: Histomorphometric findings. J. Bone Miner. Res. 1990, 5, S185–S189.
- Maeda, K.; Kobayashi, Y.; Koide, M.; Uehara, S.; Okamoto, M.; Ishihara, A.; Kayama, T.; Saito, M.; Marumo, K. The Regulation of Bone Metabolism and Disorders by Wnt Signaling. Int. J. Mol. Sci. 2019, 20, 5525.
- Day, T.F.; Guo, X.Z.; Garrett-Beal, L.; Yang, Y.Z. Wnt/β-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 2005, 8, 739–750.
- Patel, M.S.; Karsenty, G. Regulation of bone formation and vision by LRP5. N. Engl. J. Med. 2002, 346, 1572–1574.
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192.
- Chu, Y.R.; Gao, Y.H.; Yang, Y.M.; Liu, Y.; Guo, N.; Wang, L.M.; Huang, W.; Wu, L.W.; Sun, D.J.; Gu, W.K. β-catenin mediates fluoride-induced aberrant osteoblasts activity and osteogenesis. Environ. Pollut. 2020, 265, 114734.
- Karner, C.M.; Long, F.X. Wnt signaling and cellular metabolism in osteoblasts. Cell. Mol. Life Sci. 2017, 74, 1649–1657.
- Rawadi, G.; Roman-Roman, S. Wnt signaling pathway: A new target for the treatment of osteoporosis. Expert Opin. Ther. Targets 2005, 9, 1063–1077.
- Sun, D.J.; Gao, Y.H. Molecular mechanism of pathogenesis of osteofluorosis: A discussion in the view of bony turnover. Chin. J. Endemiol. 2008, 27, 239–241.
- Chen, X.S.; Yu, Y.N.; Yi, W.; Wan, L.B.; Xie, Y. Effect of fluoride on expression of mRNA and protein of Wnt3a and β-catenin in osteoblast of rats. Chin. J. Endemiol. 2013, 32, 140–145.
- Wang, W.P.; Xu, J.; Liu, K.J.; Liu, X.L.; Li, C.C.; Cui, C.Y.; Zhang, Y.Z.; Li, H.B. Suppression of Sclerostin and Dickkopf-1 levels in patients with fluorine bone injury. Environ. Toxicol. Pharmacol. 2013, 35, 402–407.
- Liu, X.L.; Li, C.C.; Liu, K.J.; Cui, C.Y.; Zhang, Y.Z.; Liu, Y. The influence of fluoride on the expression of inhibitors of Wnt/β-catenin signaling pathway in rat skin fibroblast Cells. Biol. Trace Elem. Res. 2012, 148, 117–121.
- Zeng, Q.B.; Xu, Y.Y.; Yu, X.; Yang, J.; Hong, F.; Zhang, A.H. Silencing GSK3β instead of DKK1 can inhibit osteogenic differentiation caused by co-exposure to fluoride and arsenic. Bone 2019, 123, 196–203.
- Yu, J.; Canalis, E. Notch and the regulation of osteoclast differentiation and function. Bone 2020, 138, 115474.
- Schroeter, E.H.; Kisslinger, J.A.; Kopan, R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 1998, 393, 382–386.
- Canalis, E. Notch in skeletal physiology and disease. Osteoporos. Int. 2018, 29, 2611–2621.
- Zanotti, S.; Canalis, E. Notch regulation of bone development and remodeling and related skeletal disorders. Calcif. Tissue Int. 2012, 90, 69–75.
- Hilton, M.J.; Tu, X.; Wu, X.; Bai, S.; Zhao, H.; Kobayashi, T.; Kronenberg, H.M.; Teitelbaum, S.L.; Ross, F.P.; Kopan, R.; et al. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat. Med. 2008, 14, 306–314.
- Rizzo, P.; Osipo, C.; Foreman, K.; Golde, T.; Osborne, B.; Miele, L. Rational targeting of Notch signaling in cancer. Oncogene 2008, 27, 5124–5131.
- Zanotti, S.; Canalis, E. Notch Signaling and the Skeleton. Endocr. Rev. 2016, 37, 223–253.
- Wang, S.C.; Kawashima, N.; Sakamoto, K.; Katsube, K.I.; Shindo, K.; Suda, H.; Shi, J.N. Osteogenic differentiation of murine mesenchymal progenitor cells Kusa-A1 is promoted by CBF1. Basic Clin. Med. 2006, 26, 409–414.
- Wang, S.C.; Kawashima, N.; Sakamoto, K.; Suda, H.; Shi, J.N. Expression of Notch-related Genes in the Differentiation of Mesenchymal Progenitor Cell, Kusa-A1. J. Oral Sci. Res. 2005, 21, 389–392.
- Chen, X.W.; Wan, C.W.; Xie, C.; Wei, Y.; Wu, Y.; Wan, W. Fluoride Inhibits Expressions of Notch3 and Jag1 Proteins in Rat Bone Tissues. J. Environ. Occup. Med. 2016, 33, 494–498.
- Chen, X.W.; Wan, C.W.; Xie, C.; Yang, X.X.; Wu, Y.; Wan, W. Influence of fluoride on RBPJ and related genes in bone tissue of rats. Chin. J. Public Health 2016, 32, 195–198.
- Zhang, Z.D.; Zhang, X.Z.; Zhao, D.W.; Liu, B.Y.; Wang, B.J.; Yu, W.T.; Li, J.L.; Yu, X.B.; Cao, F.; Zheng, G.S.; et al. TGF-β1 promotes the osteoinduction of human osteoblasts via the PI3K/AKT/mTOR/S6K1 signalling pathway. Mol. Med. Rep. 2019, 19, 3505–3518.
- Ma, J.; Du, D.; Liu, J.; Guo, L.; Li, Y.C.; Chen, A.; Ye, T.W. Hydrogen sulphide promotes osteoclastogenesis by inhibiting autophagy through the PI3K/AKT/mTOR pathway. J. Drug Target. 2020, 28, 176–185.
- Guan, Y.J.; Yang, X.; Yang, W.T.; Charbonneau, C.; Chen, Q. Mechanical activation of mammalian target of rapamycin pathway is required for cartilage development. FASEB J. 2014, 28, 4470–4481.
- Yu, Y.N.; Yang, D.; Zhu, H.Z.; Deng, C.N.; Guan, Z.Z. Expression of mRNA and protein of p38, Osx, PI3K and Akt1 in rat bone with chronic fluorosis. Chin. J. Pathol. 2012, 41, 622–626.
- Chen, R.; Yu, Y.N.; Xu, L.; Deng, C.N. Role of mTOR autophagy signaling in rats cartilages with fluorosis-caused damage. Chin. J. Control Endem. Dis. 2017, 32, 18–19.
- Fingar, D.C.; Salama, S.; Tsou, C.; Harlow, E.; Blenis, J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002, 16, 1472–1487.
- Zhang, R.X. Fluoride Inhibits the Proliferation and Differentiation of ATDC5 Cells via the PI3K/AKT/mTOR Signaling Pathway. Master’s Thesis, China Medical University, Shenyang, China, 2020.
- Wang, Y.; Han, C.; Lu, L.; Magliato, S.; Wu, T. Hedgehog Signaling Pathway Regulates Autophagy in Human Hepatocellular Carcinoma Cells. Hepatology 2013, 58, 995–1010.
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087.
- Long, F.X.; Zhang, X.M.; Karp, S.; Yang, Y.Z.; McMahon, A.P. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 2001, 128, 5099–5108.
- Deng, C.N. The Mechanism and Relationship of Hedgehog Signaling in the Bone Injury and Bone Microenvironment of Chronic Fluorosis Rats. Ph.D. Thesis, Guiyang Medical College, Guiyang, China, 2014.
- Gui, C.Z.; Wang, C.S.; Yu, Y.N.; Tang, J.J.; Liu, J.J. Influence of fluoride on the growth and apoptosis of cultured cartilage and antagonizing effect of superoxide dismutase (SOD). Guizhou Med. J. 2004, 28, 291–293.
- Zhang, J.Y.; Xu, S.J.; Wang, K. The Role of Free Radicals in the Pathological Process of Kaschin-Beck Disease Ⅵ. Damage of Type Ⅰ Collagen Induced by Active Oxygen Free Radicals and the Mineralization of Hydroxyapatite in the Damaced Collagen. J. Beijing Med. Univ. 1991, 23, 231–234.
- Fitch, P.M.; Howie, S.E.M.; Wallace, W.A.H. Oxidative damage and TGF-b differentially induce lung epithelial cell sonic hedgehog and tenascin-C expression: Implications for the regulation of lung remodelling in idiopathic interstitial lung disease. Int. J. Exp. Pathol. 2011, 92, 8–17.
- Zhu, Z.J.; Yu, Y.N.; Chen, R.; Huang, X.L. Role of hedgehog signaling pathway on cartilage tissue damage in chronic fluorosis rats. Chin. J. Public Health 2018, 34, 241–245.
- Datta, N.S.; Abou-Samra, A.B. PTH and PTHrP signaling in osteoblasts. Cell. Signal. 2009, 21, 1245–1254.
- Fermor, B.; Skerry, T.M. PTH/PTHrP receptor expression on osteoblasts and osteocytes but not resorbing bone surfaces in growing rats. J. Bone Miner. Res. 1995, 10, 1935–1943.
- Xu, H.; Liu, Q.Y.; Zhang, J.M.; Zhang, H.; Li, G.S. Elevation of PTH and PTHrp induced by excessive fluoride in rats on a calcium-deficient diet. Biol. Trace Elem. Res. 2010, 137, 79–87.
- Yu, X.H.; Yu, H.L.; Jiang, N.N.; Zhang, X.Y.; Zhang, M.M.; Xu, H. PTH (1-34) affects bone turnover governed by osteocytes exposed to fluoride. Toxicol. Lett. 2018, 288, 25–34.
- Neer, R.M.; Arnaud, C.D.; Zanchetta, J.R.; Prince, R.; Gaich, G.A.; Reginster, J.Y.; Hodsman, A.B.; Eriksen, E.F.; Ish-Shalom, S.; Genant, H.K.; et al. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N. Engl. J. Med. 2001, 344, 1434–1441.
- Uchida, Y.; Kuroshima, S.; Uto, Y.; Kanai, R.; Inoue, M.; Suzue, M.; Sawase, T. Intermittent administration of parathyroid hormone improves bone quality and quantity around implants in rat tibiae. J. Oral. Biosci. 2020, 62, 139–146.
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806.
- Fulzele, K.; Riddle, R.C.; DiGirolamo, D.J.; Cao, X.M.; Wan, C.; Chen, D.Q.; Faugere, M.C.; Aja, S.; Hussain, M.A.; Brüning, J.C.; et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell 2010, 142, 309–319.
- Hu, C.Y.; Ren, L.Q.; Li, X.N.; Wu, N.; Li, G.S.; Liu, Q.Y.; Xu, H. Effect of fluoride on insulin level of rats and insulin receptor expression in the MC3T3-E1 cells. Biol. Trace Elem. Res. 2012, 150, 297–305.
- Yang, C.; Zhang, M.M.; Li, Y.G.; Wang, Y.; Mao, W.X.; Gao, Y.; Xu, H. Streptozotocin Aggravated Osteopathology and Insulin Induced Osteogenesis Through Co-treatment with Fluoride. Biol. Trace Elem. Res. 2015, 168, 453–461.
- Liu, Q.Y.; Liu, H.; Yu, X.H.; Wang, Y.; Yang, C.; Xu, H. Analysis of the Role of Insulin Signaling in Bone Turnover Induced by Fluoride. Biol. Trace Elem. Res. 2016, 171, 380–390.
- Liu, C.; Zhou, X.Y.; Yang, S.R.; Li, Z.W.; Jia, Y. Mechanism and cross-talk of signaling pathways associated with bone damage in fluorosis. J. Environ. Occup. Med. 2021, 38, 794–800.
- Poole, K.E.; van Bezooijen, R.L.; Loveridge, N.; Hamersma, H.; Papapoulos, S.E.; Löwik, C.W.; Reeve, J. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005, 19, 1842–1844.
- Costa, A.G.; Bilezikian, J.P. Sclerostin: Therapeutic horizons based upon its actions. Curr. Osteoporos. Rep. 2012, 10, 64–72.
- Kramer, I.; Keller, H.; Leupin, O.; Kneissel, M. Does osteocytic SOST suppression mediate PTH bone anabolism? Trends Endocrinol. Metab. 2010, 21, 237–244.
- Gui, F.Z. Effects of Fluoride on the Expression of GAG Components and Related Signaling Pathways FGFR3 and Ihh/PTHrP in Rat Growth Plate Cartilage. Master’s Thesis, China Medical College, Shenyang, China, 2019.
- Wang, W.D. Effects of PTH and Notch Signaling Pathway on the Differentiation of Bone Mesenchymal Stem Cells into Osteoblasts. Master’s Thesis, Nanjing Medical College, Nanjing, China, 2015.
- Lin, F.T.; Xia, C.; Zhang, B.; Huang, J.G.; Zheng, X.P.; Yi, T.T.; Zhao, H.H.; Zhang, Y.B. Effects of PTHrP and Notch signaling on the proliferation of epiphysis stem cells. Natl. Med. J. China 2011, 91, 2073–2076.
- Day, T.F.; Yang, Y. Wnt and hedgehog signaling pathways in bone development. J. Bone Joint Surg. Am. 2008, 90, 19–24.
- Hu, H.; Hilton, M.J.; Tu, X.; Yu, K.; Ornitz, D.M.; Long, F. Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development 2005, 132, 49–60.
- Ng, R.C.; Matsumaru, D.; Ho, A.S.; Garcia-Barceló, M.M.; Yuan, Z.W.; Smith, D.; Kodjabachian, L.; Tam, P.K.; Yamada, G.; Lui, V.C. Dysregulation of Wnt inhibitory factor 1 (Wif1) expression resulted in aberrant Wnt-β-catenin signaling and cell death of the cloaca endoderm, and anorectal malformations. Cell Death Differ. 2014, 21, 978–989.
- Kawano, Y.; Kypta, R. Secreted antagonists of the Wnt signalling pathway. J. Cell Sci. 2003, 116, 2627–2634.
- Surmann-Schmitt, C.; Widmann, N.; Dietz, U.; Saeger, B.; Eitzinger, N.; Nakamura, Y.; Rattel, M.; Latham, R.; Hartmann, C.; von der Mark, H.; et al. Wif-1 is expressed at cartilage-mesenchyme interfaces and impedes Wnt3a-mediated inhibition of chondrogenesis. J. Cell Sci. 2009, 122, 3627–3637.
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205.
- Saidak, Z.; Le Henaff, C.; Azzi, S.; Marty, C.; Da Nascimento, S.; Sonnet, P.; Marie, P.J. Wnt/β-catenin signaling mediates osteoblast differentiation triggered by peptide-induced α5β1 integrin priming in mesenchymal skeletal cells. J. Biol. Chem. 2015, 290, 6903–6912.
- Baker, J.; Liu, J.P.; Robertson, E.J.; Efstratiadis, A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell 1993, 75, 73–82.
- Wang, T.; Zhang, X.; Bikle, D.D. Osteogenic Differentiation of Periosteal Cells During Fracture Healing. J. Cell Physiol. 2017, 232, 913–921.
- Rivas, A.; Vidal, R.L.; Hetz, C. Targeting the unfolded protein response for disease intervention. Expert Opin. Ther. Targets 2015, 19, 1203–1218.
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Invest. 2005, 115, 2656–2664.
- Moore, K.A.; Hollien, J. The unfolded protein response in secretory cell function. Annu. Rev. Genet. 2012, 46, 165–183.
- Wu, J.; Kaufman, R.J. From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ. 2006, 13, 374–384.
- Shen, X.H.; Zhang, K.Z.; Kaufman, R.J. The unfolded protein response-a stress signaling pathway of the endoplasmic reticulum. J. Chem. Neuroanat. 2004, 28, 79–92.
- Xu, H.; Jing, L.; Zhang, C.W.; Qi, L.; Li, G.S. Analysis of proteins in osteoblast exposed to fluoride by two-dimensional electrophoresis and mass spectrometry. Chin. J. Endemiol. 2006, 25, 35–38.
- Maurel, M.; Chevet, E. Endoplasmic reticulum stress signaling: The microRNA connection. Am. J. Physiol. Cell Physiol. 2013, 304, C1117–C1126.
- Mujcic, H.; Nagelkerke, A.; Rouschop, K.M.; Chung, S.; Chaudary, N.; Span, P.N.; Clarke, B.; Milosevic, M.; Sykes, J.; Hill, R.P.; et al. Hypoxic activation of the PERK/eIF2α arm of the unfolded protein response promotes metastasis through induction of LAMP3. Clin. Cancer Res. 2013, 19, 6126–6137.
- Salaroglio, I.C.; Panada, E.; Moiso, E.; Buondonno, I.; Provero, P.; Rubinstein, M.; Kopecka, J.; Riganti, C. PERK induces resistance to cell death elicited by endoplasmic reticulum stress and chemotherapy. Mol. Cancer 2017, 16, 91.
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332.
- Zhang, P.F.; Sun, Q.; Zhao, C.Y.; Ling, S.K.; Li, Q.; Chang, Y.Z.; Li, Y.X. HDAC4 protects cells from ER stress induced apoptosis through interaction with ATF4. Cell. Signal. 2014, 26, 556–563.
- Harding, H.P.; Novoa, I.; Zhang, Y.H.; Zeng, H.Q.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108.
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117.
- Xu, H.; Zhou, Y.L.; Zhang, X.Y.; Lu, P.; Li, G.S. Activation of PERK signaling through fluoride-mediated endoplasmic reticulum stress in OS732 cells. Toxicology 2010, 277, 1–5.
- Sun, F.; Li, X.N.; Yang, C.; Lv, P.; Li, G.S.; Xu, H. A role for PERK in the mechanism underlying fluoride-induced bone turnover. Toxicology 2014, 325, 52–66.
- He, P.; Zhang, M.; He, W.H.; Xia, T.; Yang, K.D.; Wang, A.G. Effects of fluoride on oxidative stress and apoptosis in primary rat hippocampal neurons. Chin. J. Endemiol. 2006, 25, 264–267.
- Shanthakumari, D.; Srinivasalu, S.; Subramanian, S. Effect of fluoride intoxication on lipidperoxidation and antioxidant status in experimental rats. Toxicology 2004, 204, 219–228.
- Xu, H.; Wang, C.H.; Zhao, Z.T.; Zhang, W.B.; Li, G.S. Role of oxidative stress in osteoblasts exposed to sodium fluoride. Biol. Trace Elem. Res. 2008, 123, 109–115.
- Shi, C.L. Protective Effect and Mechanism of Gastrodin on Rats with Chronic Fluorosis and Bone Damage. Master’s Thesis, China Medical University, Shenyang, China, 2019.
- Zhong, Y.F. The Effects of Fluoride on Nrf2-ARE Signal Pathway in Rat Osteoblasts. Master’s Thesis, Guangdong School of Pharmacy, Guangzhou, China, 2011.
- Wang, Z.; Yang, X.; Yang, S.; Ren, G.; Ferreri, M.; Su, Y.; Chen, L.; Han, B. Sodium fluoride suppress proliferation and induce apoptosis through decreased insulin-like growth factor-I expression and oxidative stress in primary cultured mouse osteoblasts. Arch. Toxicol. 2011, 85, 1407–1417.
- Park, K.H.; Park, B.; Yoon, D.S.; Kwon, S.H.; Shin, D.M.; Lee, J.W.; Lee, H.G.; Shim, J.H.; Park, J.H.; Lee, J.M. Zinc inhibits osteoclast differentiation by suppression of Ca2+-Calcineurin-NFATc1 signaling pathway. Cell Commun. Signal. 2013, 11, 74.
- Xie, Y.; Yu, Y.N.; Wan, L.B.; Chen, X.S. Effect of fluoride on expression of CaN mRNA and protein in bone tissue of rats. Chin. J. Pathol. 2012, 41, 761–764.
- Pei, J.R.; Li, B.Y.; Li, Z.W.; Wei, W.; Yao, Y.J.; Xu, J.X.; Gao, Y.H. The effect of fluoride on osteoclast in bone tissue of rats and its mechanism. Chin. J. Endemiol. 2017, 36, 714–718.
- He, H.; Liu, X.; Lv, L.; Liang, H.; Leng, B.; Zhao, D.; Zhang, Y.; Du, Z.; Chen, X.; Li, S.; et al. Calcineurin suppresses AMPK-dependent cytoprotective autophagy in cardiomyocytes under oxidative stress. Cell Death Dis. 2014, 5, e997.
- Neganova, I.; Lako, M. G1 to S phase cell cycle transition in somatic and embryonic stem cells. J. Anat. 2008, 213, 30–44.
- Zang, J.J.; Xie, F.; Xu, J.F.; Qin, Y.Y.; Shen, R.X.; Yang, J.M.; He, J. P16 gene hypermethylation and hepatocellular carcinoma: A systematic review and meta-analysis. World J. Gastroenterol. 2011, 17, 3043–3048.
- Chen, C.; Zhang, A.H.; Pan, X.L. The effects of fluoride on hypermethylation, transcription and expression of p16 gene in osteoblasts of rats. Chin. J. Endem. 2016, 35, 89–93.
- Daiwile, A.P.; Tarale, P.; Sivanesan, S.; Naoghare, P.K.; Bafana, A.; Parmar, D.; Kannan, K. Role of fluoride induced epigenetic alterations in the development of skeletal fluorosis. Ecotoxicol. Environ. Saf. 2019, 169, 410–417.
- Lv, H.H.; Tang, X.L.; Fu, S.B. Puerarin reduces methylation of estrogen receptorαpromoter in osteoblasts and regulates its proliferation and osteoblastic differentiation. J. Hebei Med. Univ. 2015, 36, 385–390.
- Zhang, Y.L.; Huang, H.; Gong, B.; Duan, L.Z.; Sun, L.; He, T.K.; Cheng, X.M.; Li, Z.Y.; Cui, L.X.; Ba, Y. Do Environmental Fluoride Exposure and ESRα Genetic Variation Modulate Methylation Modification on Bone Changes in Chinese Farmers? Chem. Res. Toxicol. 2017, 30, 1302–1308.
- Chen, T.; Liu, J. Advances in epigenetic pathogenesis of fluorosis. Chin. J. Endemiol. 2020, 39, 698–702.
- Jiang, Y.T.; Yang, Y.M.; Wang, H.G.; Darko, G.M.; Sun, D.J.; Gao, Y.H. Identification of miR-200c-3p as a major regulator of SaoS2 cells activation induced by fluoride. Chemosphere 2018, 199, 694–701.
- He, S.Y.; Chen, M.; Lin, X.L.; Lv, Z.Q.; Liang, R.Y.; Huang, L.J. Triptolide inhibits PDGF-induced proliferation of ASMCs through G0/G1 cell cycle arrest and suppression of the AKT/NF-κB/cyclinD1 signaling pathway. Eur. J. Pharmacol. 2020, 867, 172811.
- Ouyang, T.; Qin, Y.; Luo, K.K.; Han, X.; Yu, C.; Zhang, A.H.; Pan, X.L. miR-486-3p regulates CyclinD1 and promotes fluoride-induced osteoblast proliferation and activation. Environ. Toxicol. 2021, 36, 1817–1828.
- Gao, J.Y.; Qin, Y.; Luo, K.K.; Wang, X.L.; Yu, C.; Zhang, A.H.; Pan, X.L. Downregulation of miR-4755-5p promotes fluoride-induced osteoblast activation via tageting Cyclin D1. J. Trace. Elem. Med. Biol. 2020, 62, 126626.
- Luo, K.K.; Qin, Y.; Ouyang, T.; Wang, X.L.; Zhang, A.H.; Luo, P.; Pan, X.L. Let-7c-5p Regulates CyclinD1 in Fluoride-Mediated Osteoblast Proliferation and Activation. Toxicol. Sci. 2021, 182, 275–287.
- Kapinas, K.; Kessler, C.; Ricks, T.; Gronowicz, G.; Delany, A.M. miR-29 modulates Wnt signaling in human osteoblasts through a positive feedback loop. J. Biol. Chem. 2010, 285, 25221–25231.
- Kapinas, K.; Kessler, C.B.; Delany, A.M. miR-29 suppression of osteonectin in osteoblasts: Regulation during differentiation and by canonical Wnt signaling. J. Cell Biochem. 2009, 108, 216–224.
- Wang, T.; Xu, Z. miR-27 promotes osteoblast differentiation by modulating Wnt signaling. Biochem. Biophys. Res. Commun. 2010, 402, 186–189.
- Yang, C.; Wang, Y.; Xu, H. Treatment and Prevention of Skeletal Fluorosis. Biomed. Environ. Sci. 2017, 30, 147–149.
- Wang, W.Y.; Gui, C.Z.; Guan, Z.Z. Research progress on antagonists of fluorosis. Occup. Health 2021, 37, 2433–2438.
- Gupta, S.K.; Gupta, R.C.; Seth, A.K.; Gupta, A. Reversal of fluorosis in children. Acta Paediatr. Jpn. 1996, 38, 513–519.
- Guo, S.Q.; Yu, M.J.; Shen, H.P.; Wang, D.; Yuan, Z.H.; Cheng, J.F. Histomorphometry Effect of Compound Traditional Chinese Medicine on Rats Skeletal Fluorosis. Mod. Prev. Med. 2016, 43, 1471–1475.
This entry is offline, you can click here to edit this entry!