Inner ear malformations are a spectrum of congenital anomalies involving the inner ear structures with an emphasis on the cochlea due to their implications for sensorineural hearing loss.
- inner ear malformations
- hearing loss
- genetics
- complete labyrinthine aplasia
- common cavity
1. Introduction
Inner ear malformations are estimated to be present in around 20% of patients with congenital sensorineural hearing loss [1].
The relationship between inner ear anomalies and hearing impairment has been known since the first anatomical report in 1791 by Carlo Mondini, of Italy, and many distinct malformations have been described in the last 230 years [2].
In recent years, the increasing availability of genetic consultation and testing has provided new data about the possible connection between inner ear malformations and genetic anomalies. In addition, the evolution of neuroradiological imaging with new computed tomography (CT) methods and magnetic resonance imaging (MRI) provided new data about syndromic patients. Indeed, in some syndromes, the well-known presence of hearing loss has been associated with the presence of inner ear malformations, in some cases extremely specific ones.
Table 1 summarizes the current understanding about the genetics of inner ear malformations
| Type of Malformation | Possible Related Syndrome | Genes Involved |
|---|---|---|
| Complete labyrinthine aplasia | Wildervanck syndrome | - |
| LAMM syndrome | FGF3 | |
| HOXA1 mutation syndrome | HOXA1 | |
| Rudimentary otocyst | - | - |
| Common cavity | Bosley–Salih–Alorainy syndrome | HOXA1 |
| 22q11 deletion syndrome | - | |
| - | ROR1 | |
| Cochlear aplasia | - | PAX2 (animal studies) |
| Incomplete partition type 1 | - | - |
| Incomplete partition type 2 | Pendred syndrome | SLC26A4 |
| - | FOXI1 | |
| - | KCNJ10 | |
| Incomplete partition type 3 | - | POUF3F4 |
| Cochlear hypoplasia | Branchio-oto-renal syndrome | - |
| Oculo-auriculo-vertebral spectrum disorder | - | |
| Warsaw breakage syndrome | DDX11 | |
| Pallister–Hall syndrome | - | |
| - | SIX1 | |
| - | CHD7 (animal studies) | |
| - | TBX2 (animal studies) | |
| - | HOXA1/HOXB1 (animal studies) | |
| Cochlear hypoplasia Type 1 | - | - |
| Cochlear hypoplasia Type 2 | - | - |
| Cochlear hypoplasia Type 3 | Waardenburg syndrome | SOX10 |
| Cochlear hypoplasia Type 4 | ⲁ-Dystroglycan-related muscular disorders (Walker–Warburg syndrome) | - |
| Posterior labyrinth anomalies | CHARGE syndrome | - |
| Trisomy 13 | - | |
| Trisomy 18 | - | |
| Waardenburg syndrome type II | - | |
| Alagille syndrome | JAG1 | |
| Warsaw breakage syndrome | - |
2. Cochlear Aplasia (with and without a Dilated Vestibule)
Cochlear aplasia is characterized by a complete absence of the cochlea, with absence of the cochlear nerve canal and cochlear nerve, and it is supposed to account for only 3% of cochlear malformations (see Figure 1 ) [3].
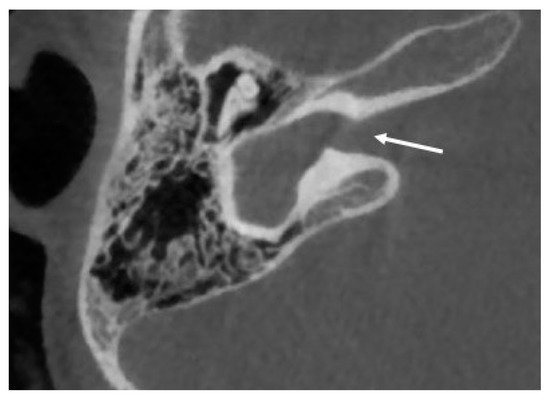
Two subtypes can be identified of this anomaly: with and without dilated vestibule [1]. This malformation causes a profound sensorineural hearing loss. The possibility of cochlear implantation is controversial.
No specific gene mutations responsible for cochlear aplasia are reported in the literature, but PAX genes were supposedly involved in the development of the inner ear from experimental studies on animal models, in particular anomalies in gene PAX2 [4].
3. Cochlear Hypoplasia
The current classification system [1][5] accounts for four subtypes of cochlear hypoplasia, previously described in numerous studies: (1) cochlear hypoplasia type 1: small budlike cochlea without an internal architecture; (2) cochlear hypoplasia type 2: a cochlea with an architecture similar to normal, reduced in size, with a modiolus or interscalar septa being present but defective; (3) cochlear hypoplasia type 3: internal and external architecture quite normal, but the size is reduced with fewer or shorter turns; and (4) cochlear hypoplasia type 4: a cochlea reduced in size with a normal basal turn and hypoplasia of the middle and apical turns [6][7].
Even if this classification describes four different subtypes of cochlea hypoplasia, most articles available in the literature categorize these anomalies with the general term of “cochlear hypoplasia” without further characterization. The above-mentioned limit explains why the connection between cochlear hypoplasia and genetic anomalies is difficult, and the relation between cochlear hypoplasia subtypes and genetic anomalies is currently impossible to describe.
Recently, a de novo missense variant [p(Asn174Tyr)] in the DNA-binding homeodomain of SIX1 was identified in a single patient with bilateral hearing loss due to cochlear hypoplasia and cochleovestibular nerve abnormality. This gene was previously associated with autosomal dominant hearing loss and branchio-oto-renal or branchio-otic syndrome [8].
Studies conducted on mice showed that mutations in CHD7, a gene mutated in human CHARGE syndrome, lead to cochlear hypoplasia [9], while humans with CHARGE syndrome frequently present different anterior [10] and posterior labyrinth anomalies [11]. In addition, the ablation of TBX2 from the otocyst leads to cochlear hypoplasia in mouse models [12].
4. Posterior Labyrinth Anomalies
The abnormalities of the posterior labyrinth are a wide spectrum of anomalies not fully characterized yet. The anterior labyrinth has always captured most of the attention because of the impact of a congenital hearing loss in the neuro-psycho-social evolution of human beings. On the other hand, motor and balance development seems to be less affected by congenital anomalies of the posterior part of the labyrinth because of the possible compensatory role of the visual and neurological systems. So, the role of these anomalies is still under evaluation.
Dysplasia of the semicircular canals, isolated or combined with cochlear anomalies, is described in several syndromes such as CHARGE syndrome, trisomy 13, and trisomy 18 [13]. In CHARGE syndrome it is typical to detect abnormalities in all the semicircular canals [14]. On the other hand, a partial involvement can be observed in trisomy 13, in which lateral semicircular canal defects are most common and are present with or without superior or posterior semicircular canal abnormalities, while in trisomy 18, the defects frequently affect the lateral and superior semicircular canals [15]. Isolated agenesis of the posterior semicircular canals, without involvement of the lateral semicircular canals, is extremely rare but has been detected in Waardenburg syndrome type II patients [16].
Bilateral anomalies of the posterior semicircular canals, with frequent concomitant involvement of the superior semicircular canals but normal lateral semicircular canals, were also described by Okuno in 1990 in the temporal bones of four individuals with Alagille syndrome [17]. This condition is characterized by chronic cholestasis, characteristic facial appearance, cardiovascular abnormalities, vertebral arch defect, growth retardation, mental retardation, and hypogonadism [18]. This syndrome is caused by mutations in the JAG1 gene [19]. According to Koch et al. the observation of a similar condition in Waardenburg syndrome type II suggests that this gene may have a crucial role for the development of the posterior semicircular canal, but not for the lateral semicircular canal that has a later development [13]. In some patients with Warsaw breakage syndrome, semicircular canal anomalies have been detected [20].
This entry is adapted from the peer-reviewed paper 10.3390/audiolres11040047
References
- Sennaroğlu, L.; Bajin, M.D. Classification and Current Management of Inner Ear Malformations. Balk. Med. J. 2017, 34, 397–411.
- Brotto, D.; Uberti, A.; Manara, R. From Mondini to the latest inner ear malformations’ classifications: An historical and critical review. Hear. Balance Commun. 2019, 17, 241–248.
- Joshi, V.M.; Navlekar, S.K.; Kishore, G.R.; Reddy, K.J.; Kumar, E.C. CT and MR imaging of the inner ear and brain in children with congenital sensorineural hearing loss. Radiographics 2012, 32, 683–698.
- Bouchard, M.; de Caprona, D.; Busslinger, M.; Xu, P.; Fritzsch, B. Pax2 and Pax8 cooperate in mouse inner ear morphogenesis and innervation. BMC Dev. Biol. 2010, 10, 89.
- Sennaroglu, L. Histopathology of inner ear malformations: Do we have enough evidence to explain pathophysiology? Cochlea Implant. Int. 2015, 17, 3–20.
- D’Arco, F.; Sanverdi, E.; O’Brien, W.T.; Taranath, A.; Talenti, G.; Blaser, S.I. The link between inner ear malformations and the rest of the body: What we know so far about genetic, imaging and histology. Neuroradiology 2020, 62, 539–544.
- Talenti, G.; Manara, R.; Brotto, D.; D’Arco, F. High-resolution 3 T magnetic resonance findings in cochlear hypoplasias and incomplete partition anomalies: A pictorial essay. Br. J. Radiol. 2018, 91, 2018120.
- Kari, E.; Llaci, L.; Go, J.L.; Naymik, M.; Knowles, J.A.; Leal, S.M.; Rangasamy, S.; Huentelman, M.J.; Friedman, R.A.; Schrauwen, I. A de novo SIX1 variant in a patient with a rare nonsyndromic cochleovestibular nerve abnormality, cochlear hypoplasia, and bilateral sensorineural hearing loss. Mol. Genet. Genom. Med. 2019, 7, e995.
- Hurd, E.A.; Poucher, H.K.; Cheng, K.; Raphael, Y.; Martin, D.M. The ATP-dependent chromatin remodeling enzyme CHD7 regulates pro-neural gene expression and neurogenesis in the inner ear. Development 2010, 137, 3139–3150.
- Brotto, D.; Manara, R.; Gallo, S.; Sorrentino, F.; Bovo, R.; Trevisi, P.; Martini, A. Comments on “hearing restoration in cochlear nerve deficiency: The choice between cochlear implant or auditory brainstem implant, a meta-analysis”. Otol. Neurotol. 2019, 40, 543–544.
- D’Arco, F.; Youssef, A.; Ioannidou, E.; Bisdas, S.; Pinelli, L.; Caro-Dominguez, P.; Nash, R.; Siddiqui, A.; Talenti, G. Temporal bone and intracranial abnormalities in syndromic causes of hearing loss: An updated guide. Eur. J. Radiol. 2020, 123, 108803.
- Kaiser, M.; Wojahn, I.; Rudat, C.; Lüdtke, T.H.; Christoffels, V.M.; Moon, A.; Kispert, A.; Trowe, M.O. Regulation of otocyst patterning by Tbx2 and Tbx3 is required for inner ear morphogenesis in the mouse. Development 2021, 148.
- Koch, B.; Goold, A.; Egelhoff, J.; Benton, C. Partial absence of the posterior semicircular canal in Alagille syndrome: CT findings. Pediatr. Radiol. 2006, 36, 977–979.
- Guyot, J.P.; Gacek, R.R.; DiRaddo, P. The temporal bone anomaly in CHARGE association. Arch. Otolaryngol. Head Neck Surg. 1987, 113, 321–324.
- Sando, I.; Bergstrom, L.; Wood, R.P., 2nd; Hemenway, W.G. Temporal bone findings in trisomy 18 syndrome. Arch. Otolaryngol. Head Neck Surg. 1970, 91, 552–559.
- Higashi, K.; Matsuki, C.; Sarashina, N. Aplasia of posterior semicircular canal in Waardenburg syndrome type II. J. Otolaryngol. 1992, 21, 262–264.
- Okuno, T.; Takahashi, H.; Shibahara, Y.; Hashida, Y.; Sando, I. Temporal bone histopathologic findings in Alagille’s syndrome. Arch. Otolaryngol. Head Neck Surg. 1990, 116, 217–220.
- Alagille, D.; Odièvre, M.; Gautier, M.; Dommergues, J.P. Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental, and sexual development, and cardiac murmur. J. Pediatr. 1975, 86, 63–71.
- Li, L.; Krantz, I.D.; Deng, Y.; Genin, A.; Banta, A.B.; Collins, C.C.; Qi, M.; Trask, B.J.; Kuo, W.L.; Cochran, J.; et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat. Genet. 1997, 16, 243–251.
- Alkhunaizi, E.; Shaheen, R.; Bharti, S.K.; Joseph-George, A.M.; Chong, K.; Abdel-Salam, G.M.H.; Alowain, M.; Blaser, S.I.; Papsin, B.C.; Butt, M.; et al. Warsaw breakage syndrome: Further clinical and genetic delineation. Am. J. Med. Genet. Part A 2018, 176, 2404–2418.