Hepatitis B virus (HBV) is a small enveloped DNA virus which replicates its tiny 3.2 kb genome by reverse transcription inside an icosahedral nucleocapsid, formed by a single ~180 amino acid capsid, or core, protein (Cp). HBV causes chronic hepatitis B (CHB), a severe liver disease responsible for nearly a million deaths each year. Dynamic changes in Cp chemical modification and capsid conformation are crucial in the viral life-cycle and represent a promising new antiviral target.
- capsid assembly modulator (CAM)
- chronic hepatitis B (CHB)
- core protein
- HBV
- HBV cure
1. Introduction
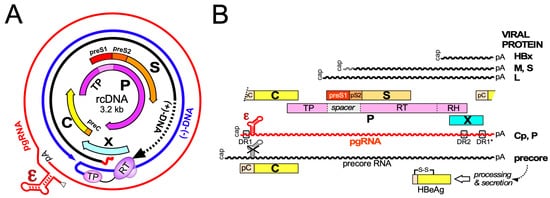
Hepatitis B virus (HBV), the etiological agent of acute and chronic hepatitis B (CHB) in humans, is a hepatotropic small enveloped DNA virus that replicates through reverse transcription. Although its ~3 kb genome encodes only seven primary gene products ( Figure 1 ) HBV is one of the most successful human pathogens. According to World Health Organization (WHO) estimates two billion people carry antibodies indicating prior exposure to the virus, and close to 300 million have become chronic virus carriers [1]. They are at a high risk to develop severe liver disease such as fibrosis, cirrhosis, and primary liver cancer [2], with nearly a million deaths per year. HBV infection can be prevented by a prophylactic vaccine yet current treatments for chronic infection are usually not curative [3]. Owing to pronounced adverse effects, only a small fraction of patients are eligible for type-I interferon (IFN) based therapies, e.g., with pegylated IFN-α (pegIFNα), which after a finite 24- or 48-week treatment leads in ~10% of the patients to a sustained loss of hepatitis B surface antigen (HBsAg) [3]. Often the virus is controlled but not eliminated, a condition termed “functional cure”. Most patients are instead treated with one of the six FDA approved and better tolerated nucleos(t)ide analogs (NUCs) which inhibit reverse transcription by the multidomain HBV polymerase (P protein).
Early NUCs were prone to rapid viral resistance development, for lamivudine in up to 70% of patients after four years of treatment [4]. The current first-line NUCs entecavir, tenofovir disoproxil fumarate (TDF), and tenofovir alafenamide (TAF) are more potent and resistance rates are at most 1% after several years of treatment [5]. The high resistance barrier results from the massive suppression of viral genome replication, with >10 6 fold declines in serum HBV levels, and the dependence on multiple mutations for the evolution of drug-resistant yet reasonably active enzyme P protein. Still, long-term (probably life-long) NUC therapy is necessary for most patients because, despite dampened liver inflammation, HBsAg seroclearance is rare. This is in part due to mRNA transcription from chromosomally integrated HBV DNA [6], a by-product of, but not essential for, the viral replication cycle, yet mainly to the NUCs not targeting the viral persistence reservoir, the covalently closed circular DNA (cccDNA) form of the viral genome [7]; cccDNA is produced in the host cell nucleus from the relaxed circular (rc) DNA present in infectious HB virions and is the template proper for transcription of the viral RNAs by host RNA polymerase II (see below). Hence, although formation of rcDNA containing nucleocapsids is largely blocked by NUCs, viral antigens, and immature RNA-containing nucleocapsids continue to be produced. Hence, upon therapy cessation virus replication can fully resume. As cccDNA may persist for decades, HBV reactivation can even occur long after a past self-limited acute hepatitis B [8] when immune control is lost by an unrelated disease or upon immunosuppressive treatment.
Global efforts have therefore been initiated towards new curative treatments for chronic hepatitis B, to achieve the sustained suppression of viral replication after treatment cessation, i.e., functional cure, and, ideally, complete elimination of all HBV genomes from the body [9], with several intermediate distinctions [10]. Various innovative therapeutic approaches are pursued [11] which can be divided into three main categories, namely, direct acting antivirals (DAAs) targeting viral factors [12][13], inhibitors of HBV-relevant host factors [14] including the entry receptor NTCP (see below), and immune activation, aiming to restore an adequate immune response against the virus, exhaustion of which is a hallmark of CHB [2][15][16][17]. Amongst the most advanced new DAAs are the capsid assembly modulators (CAMs), also called core protein allosteric modulators (CpAMs), capsid assembly effectors (CAEs), or, very simplifying, capsid inhibitors (CIs), which target the capsid-forming HBV core protein (Cp); they are in the focus of this review. Even though the phenotypic consequences of different CAMs may differ, their common mechanism of action (MoA) is to disable the proper dynamics of Cp and the capsid.
Excellent recent reviews on the general concept of therapeutically targeting viral structural proteins [18], analytical methods to monitor drug activities [19], and on specific CAM chemotypes [20] are available. Here we intend to provide a comprehensive and comprehensible overview on the functional dynamics of HBV Cp in the viral life-cycle, on the basics of the underlying biochemical and structural dynamics, and on up-to-date clinical data on the therapeutic use of CAMs against CHB.
2. Functional Dynamics of the HBV Core Protein and Capsid in Virus Replication
The notion of viral capsid proteins is largely shaped by the highly symmetric capsid structures they can form, classically determined by X-ray crystallography (see below). Capsids provide a virus with a stable container for its genome to travel through space and time, and many structural biology techniques rely as well on the stability of these particles. However, a look on the role of capsid proteins in viral replication in general, and that of the HBV Cp in particular, reveals a highly dynamic behavior which is crucial for maintaining the viral life cycle.
Chromatinization of cccDNA by cellular histone and non-histone proteins plus viral proteins enable its regulated use as transcription template for new 5′ capped and 3′ polyadenylated viral RNAs by cellular RNA polymerase II. Eliminating cccDNA would thus truly cure infection, a highly ambitious goal [21]. Easier to realize appears to silence transcription from cccDNA which depends on the viral HBx protein. A major function of HBx is to mediate ubiquitylation and subsequent proteasomal degradation of the cellular structural maintenance of chromosomes 5/6 complex (SMC5/6) which suppresses cccDNA transcription [22]. As part of this mechanism, or in addition, HBx seems to govern intranuclear cccDNA localization which in turn contributes to its transcriptional activity [23][24]. HBx and its interactions with cellular factors have thus also become new targets for therapeutic intervention [25][26].
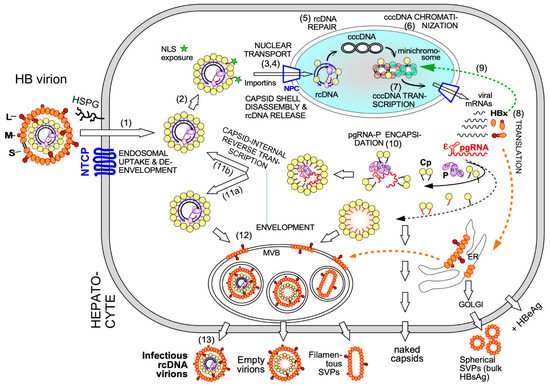
Electron-microscopy [27] and, as for HBx [28], chromatin immunoprecipitation (ChIP) data suggest that also some Cp is associated with cccDNA, raising the possibility that Cp contributes as well to cccDNA stability and/or transcriptional activity. A nuclear Cp interactome study identified numerous cellular RNA binding proteins, amongst them serine- and arginine-rich splicing factor 10 (SRSF10) which suppresses HBV transcription [29]; the interaction with Cp may counteract such a restriction. Conversely, Cp has been proposed to recruit cellular restriction factors of the APOBEC family to cccDNA, inducing its (partial) degradation [30]; in this view, maintaining a long-term association of Cp with cccDNA [31] could even benefit APOBEC inducing antiviral approaches. More work will be required to firmly establish cccDNA regulatory activities for Cp; it should as well be considered that the rcDNA entering the nucleus comes associated with Cp, hence ChIP assays may detect residual Cp that escaped replacement by chromatin components. Along this line, recent studies have shown that a mutant HBV defective for Cp production (itself produced by trans-complementation with a Cp expression vector) is still infectious and able to generate transcriptionally active cccDNA [32] which remained stable for over two months [33]. This also argues that the intracellular recycling pathway (see Figure 2 step 11b), supposedly dependent on de novo Cp synthesis and highly active for DHBV [34][35], has only a minor role for HBV cccDNA replenishment, stability, or transcriptional activity, at least in this model. Blocking recycling might then not be very effective in reducing cccDNA levels. For DHBV the more active recycling and higher cccDNA copy numbers per cell may compensate the lack of a transcription-activating HBx-like gene product, possibly via extra functions of the larger avian HBV Cp [36]. Notably, other studies on HBV concluded that stable cccDNA levels are maintained by both genome recycling and de novo secondary infections [37]. Both mechanisms would support the rapid emergence of resistant viruses during therapy with early NUCs [4][38], either by adding the mutant cccDNA to the resident wild-type (wt) cccDNA in the nucleus, or by de novo deposition in naīve cells of only mutant cccDNA. Seemingly an academic issue, determining the relative contributions of the two cccDNA pathways is crucial for future therapy design [21]. If cccDNA is mostly replenished by de novo infection, entry blockers and, possibly, also CAMs targeting the incoming nucleocapsid could effectively drain the existing cccDNA pool but would be ineffective against intracellular cccDNA recycling. The reverse holds if recycling was the dominant pathway to cccDNA maintenance.
Clearly, however, the crucial initial event is specific binding of P protein to the 5′ proximal ε stem-loop structure ( Figure 2 and Figure 3 A) on pgRNA (though not the precore RNA; [39]), with the resulting ribonucleoprotein (RNP) complex nucleating capsid shell assembly [40] by a still ill-defined mechanism.
3. Overall Structural Dynamics of HBV Cp
While X-ray studies can achieve resolutions down to below 1.5Å, the length of a covalent C-C bond, a major drawback is the need for crystallized samples which, in turn, is hampered by sample heterogeneity, including T = 3 vs. T = 4 dimorphism (see above). In cryo-EM, by contrast, single particles on the micrographs can be selected for image reconstruction. Averaging thousands of such particles then yields higher-resolution information, initially down to about 4Å [41], where the protein backbone and bulky sidechains became visible. The subsequent “resolution revolution” [42] brought by new detectors pushed this limit to below 3Å which reveals many, including smaller, sidechains. The currently highest resolution cryo-EM derived model for HBV capsids from full-length Cp183 (PDB 6HTX) has a nominal resolution of 2.66Å [43] but even higher resolution is principally possible [44]. Hence cryo-EM and cryo-electron tomography (cryo-ET), which generates multiple views of the same particles from different angles by specimen tilting, have become a mainstay for the study of virus structure [45].
X-ray crystallography and cryo-EM rely on sampling many identical particles; heterogeneity in the specimen will eventually cause a loss of resolution. NMR can monitor changes in the vicinity of NMR-active nuclei over a broad range of timescales [46] but is also an “ensemble technique” that reports on the average properties of a population of molecules or particles. Various less commonly known techniques are highly useful, especially in combination, to follow assembly some of which we will briefly address; for more information see reference [19].
A basic optical technique is light scattering which increases with particle size. Several variations exist, recently also for single-particle imaging, similar to fluorescence correlation spectroscopy. Mass spectrometry (MS) can also offer single-particle resolution, and especially charge-detection MS (CDMS) has been used to identify numerous small transitory as well as more stable “late-stage” Cp assembly intermediates, with masses between those of T = 3 and T = 4 HBV capsids [47]. Another advance are microfluidic and nanopore-based techniques, such as resistive-pulse sensing where particles of different sizes passing through the pore cause correlating changes in conductivity. Atomic force microscopy (AFM) can provide additional information on the mechanical properties of particles, including how they are affected by the material inside the particle, e.g., nucleic acid. Not the least, theoretical approaches such as molecular dynamics (MD) simulations can now handle an entire capsid [48]. A recent all-atom MD simulation of HBV capsids bound with a CAM compound suggested a mechanism for crosstalk between intra- and inter-dimer interfaces and thus a way how allostery can traverse through the entire Cp dimer [49] that is in line with experimental data [50]. Another finding was a population of dimers with significantly splayed intra-dimer interface; such opening of the four-helix bundle has also been seen in experimental studies, e.g., when capsids interact with spike-binding peptides [51], or by mutation of the spike tip-located D78 residue which, again, affects the rate of assembly [52]. While the biological meaning is not yet resolved, these convergent data exemplify how different approaches can eventually point into the same direction. Combining the different techniques can therefore provide unprecedented new insights into HBV capsid assembly as well as its inhibition; for instance, owing to the crosstalk throughout the dimer, compounds acting on the intradimer interface or the spike tip could similarly act as allosteric assembly modulators as the currently dominating CAMs that target the interdimer interface.
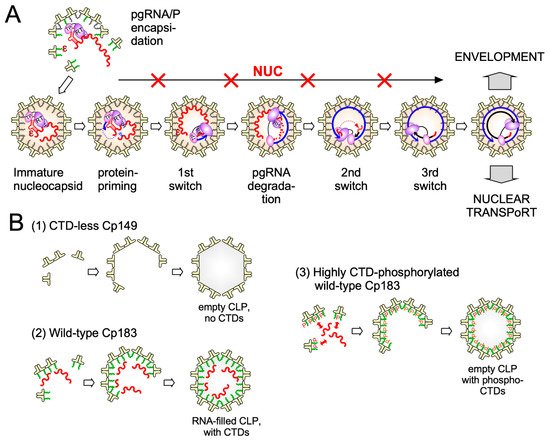
While new technologies promise much more detailed insights into HBV capsid assembly some basic aspects of the pathway are already established. Assembly proceeds most likely by a nucleation mechanism (comparable to the induction of crystallization from an oversaturated solution when a “crystal seed” is added), i.e., the rate-limiting assembly step is the formation of an intermediate (the nucleation seed) to which additional dimeric subunits can be added (see Figure 3 ), in the elongation phase, on a downhill energetic path [53]. The likely nucleation seed for neat CTD-less Cp derivatives such as Cp149 is a three-fold symmetric trimer of dimers [54], as supported by recent EM evidence [55]; this study identified, in addition, a two-fold symmetric pentamer as another predominant intermediate; there, two more dimers are associated to a trimer of dimers so as to generate two adjacent triangles. Considering that each Cp dimer is tetravalent ( Figure 4) the trigonal trimer of dimers saturates 6 of the 12 valencies, the two-fold symmetric pentamer 12 of the totally 20 valencies; hence these arrangements minimize the number of free valencies. As assembly proceeds, the percentage of free valencies per subunit decreases further until all valencies are saturated upon addition of the last dimer. In turn, removing the first subunit from a fully assembled capsid has the highest energy cost [18], hence assembly and disassembly can be nonsymmetrical processes showing hysteresis [56] ; sophisticated techniques such as time-resolved small angle X-ray scattering confirm this view [57]. Moreover, assembly may proceed via more than one pathway, as indicated by the observation with single-particle techniques such as CDMS of particles with fewer (“defective”) or more subunits (“overgrown”) than those defining a perfect icosahedron. Both defects can slowly be corrected, suggesting that completion is a separate phase in assembly [58]. Also a recent single particle cryo-EM study found defective particles to account for a substantial fraction of the total population, perhaps reflecting that nature can live with, or even exploits, imperfection in capsid assembly [59]. The actual assembly conditions are also important for the pathways; mild conditions (including low Cp concentration and ionic strength) favor regular T = 4 CLP formation with few tangible intermediates, aggressive conditions (including high Cp concentration and ionic strength) favor formation of larger, kinetically trapped intermediates and a higher proportion of T = 3 capsids. However, the correlation of any of these in vitro conditions with assembly of replication-competent HBV nucleocapsids remains to be examined.
4. Targeting HBV Capsid Dynamics
The two major CAM impacts on HBV nucleocapsid assembly, initially largely based on negative staining EM, are the induction of non-capsid-like aberrant multimers by CAM-A compounds, and the formation of apparently regularly shaped but genome-less capsids by CAM-E compounds; a prototypic CAM-A is the HAP BAY41-4109, a prototypic CAM-E the phenylpropenamide AT-130.
Higher resolution structural studies of CLPs with bound CAMs provided more direct information. The assumed tolerance of the capsid structure towards CAM-E compounds suggested by ensemble measurements was challenged by single particle analysis which revealed that AT-130 induced formation of empty as well as only partially completed particles [60]. In vivo such holes would dramatically change accessibility of the capsid interior to macromolecules, amongst them kinases and phosphatases, nucleases, or components of the innate immune system, all of which might represent additional layers of antiviral activity.
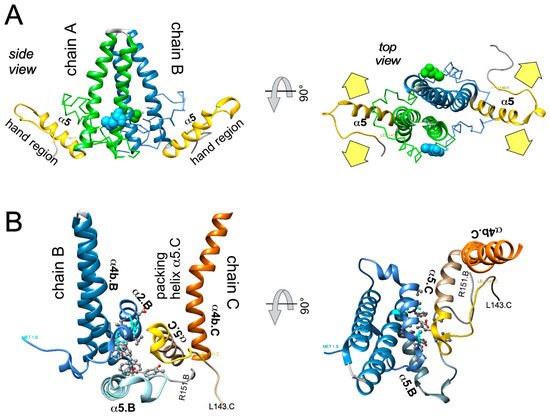
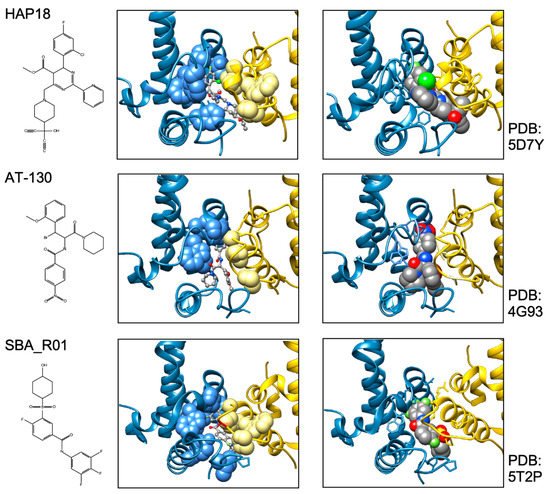
In sum do the biophysical data show that all current CAMs bind to essentially the same pocket at the interdimer interface; a comprehensive list of available high-resolution HBV capsid and Cp Y132A hexamer structures with bound CAMs is given in ref. [20]. They all locally stabilize the interaction and accelerate assembly, but, depending on the detailed binding mode, they can have differential impacts on the capsid structure and stability. For classic CAM-E compounds the kinetic effects seem to dominate over structural perturbations, resulting in seemingly normal capsids but overruling the role of the pgRNA-P protein complex as the natural nucleation seed (“over-initiation”). Conversely, for classic CAM-A compounds structural perturbation appears more important. However, with the discovery of ever more CAM chemotypes and derivatives thereof a whole spectrum of phenotypes can be expected which will also depend on drug concentration and binding site occupancy. All these effects should interfere with proper progression of the viral lifecycle.
Numerous CAM-A and CAM-E compounds made it into clinical trials; only a few are mentioned here. GLS4, a direct HAP successor of BAY41-4109, had initially shown less pronounced anti-HBV activity than other CAMs which could be related to its metabolization by CYP3A, one of the cytochrome P450 isoenzymes. Co-application of the CYP inhibitor ritonavir (RTV) enhanced GLS4 trough concentrations and boosted antiviral activity in phase 2 studies, with reported up to 4.4 log10 reductions in HBV DNA without added NUC [61]. Ongoing phase 2b studies evaluate the combination of GLS4/RTV with NUC (entecavir); interim data indicate also here superiority over NUC alone, with transient ALT flares correlating with stronger antigen declines [61]. Another CAM-A HAP compound, RO7049389, also achieved up to > 3 log10 declines in HBV DNA after 28 days of treatment in phase 1 [62][63], and entered phase 2 trials evaluating long-term triple combinations, including with investigational drugs. A further drug in phase 2 is QL-007 about which little detail has been disclosed; notably, a phase 2 study of ABI-H2158, a more potent CAM-E than ABI-H0731, plus NUC has just been discontinued following elevated ALT levels in some patients ( https://investor.assemblybio.com/news-releases/news-release-details/assembly-bio-announces-decision-discontinue-clinical-development ; accessed on 21 September 2021).
All direct-acting anti-infectives are prone to resistance development which also plagued early anti-HBV RT inhibitors [4]. Host-factor targeting therapies circumvent this problem but are more prone to exert adverse effects [14]. Resistance evolution requires a certain mutation rate and the production of a sufficient number of progeny genomes to generate, select, and fix the proper mutation(s) in the population. Hence a high resistance barrier relies on efficient suppression of genome replication, and a high number of mutations required to achieve resistance while not losing function. This explains the success of entecavir and tenofovir as anti-HBV NUCs. The same criteria hold for CAMs. Regarding replication suppression most CAMs still lag behind NUCs, hence elucidating the Cp sequence space that fits resistance plus proper nucleocapsid functioning is a crucial issue. As propagating HBV in the lab is still not feasible one way to address this question is structure-guided mutagenesis followed by characterization of the respective virus variants in cell culture, the other information source are sequencing data from clinical trials. The high-resolution structures of capsids with bound CAMs, as for instance those in Figure 5, provide detailed information on the Cp residues involved in CAM binding, and various mutational studies have functionally confirmed their relevance for resistance. For BAY41-4109 up to ~50-fold increased EC 50 values regarding HBV DNA reduction were observed for the pocket mutants D29G, Y118F and especially T33N which retained about one third the replication capacity of wt-HBV [64]. The similarity but non-identity of binding modes of different CAMs ( Figure 5 ) is also reflected in their resistance profiles; for instance, P25A/S and V124F reduced susceptibility to HAP_R01 but not to the sulfamoylbenzamide SBA_R01 [65]. A new phenotyping assay facilitating comparative analyses of more mutants yielded similar results, indicating that Cp positions 33, 102, 118, and 127 affect both HAP and SBA binding while positions 25 and 109 are more important for HAP than SBA binding [66]. Also here mutation T33N conferred the highest resistance against both CAM chemotypes yet retained about half the replication capacity of wt-HBV. A comprehensive recent study probed 25 HAP pocket residues by 70 single-site substitutions for functionality and resistance to various CAM phenotypes [67]. Most replacements of W102, interacting with many CAMs, abolished Cp assembly; Cp W102Y and Cp W102H formed capsids but exerted much reduced pgRNA encapsidation and viral DNA synthesis. Hence the emergence of single-site resistance at this position, and similarly at Y132, is unlikely. Position T33 tolerated only substitutions with chemically similar residues but these include T33N. Overall, the most relevant resistance conferring mutations identified concerned position P25, T33, I105, and S106. More such studies will help to define potential escape routes for the virus and, specifically, monitor patients for dangerous variants. These will likely not be restricted to single-site mutants because, given the opportunity, even variants with severely hampered replication capacity can regain fitness by compensatory second-site mutations, as is well known from NUC resistance [4]. Notably, natural polymorphisms have been found at HAP pocket positions by searches in HBV databases [68] and a large patient cohort [66], including Y118F and D29A, I105T/L/V and T114I/V but not T33N ; the presence or absence of such baseline mutations may thus affect treatment outcome in patients, as seen by 20-fold reduced activity to AB-506 to Cp baseline mutant I105T [69]. In an early 28-day trial with JNJ-6379 half of the patients carried viruses with one or more potentially relevant mutations, including Y118F, I105T, T109M, and T019I, but their therapy response was not generally reduced, despite a detectable enrichment of the Y118F mutant [70]. Importantly, though, 24 week interim data from the JNJ-6379 monotherapy arm of the 48 week JADE study revealed viral breakthrough in several patients which correlated with baseline mutations Y118F and I105T and emergence of the T33N mutation [71]. By contrast, no viral breakthrough or enrichment of CAM resistant variants occurred in the patients from the CAM plus NUC arm of the study. Similar data were seen with ABI-H0731 monotherapy [72] versus a one-year combination treatment with ABI-H0731 plus NUC [73], corroborating the resistance-suppressing effect of more efficient replication inhibition by the drug combination.
5. Conclusions and Perspectives
Meanwhile small molecules targeting HBV nucleocapsid assembly have proven their ability to interfere with HBV replication in clinical settings. The efficacy of viral DNA suppression by advanced CAMs is approaching that of current first-line NUCs, hence capsid assembly modulation is clearly a valid antiviral strategy. However, as with NUCs viral rebound after cessation of CAM therapy seems common. Hence present CAMs provide a highly valuable addition but not a fundamentally superior substitute for current NUC monotherapy. Nonetheless, prospects for significant improvements are good even though more virological, pharmaceutical and clinical efforts will be needed for a final judgement. Detailed knowledge on the impact of CAMs as well as the other new treatment options on the virus, the cell, and the host organism will be the best investment for devising smart combination therapies which currently hold the greatest promise for the long-awaited HBV cure.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines9111577
References
- WHO. Hepatitis B Fact Sheet. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b (accessed on 21 September 2021).
- Iannacone, M.; Guidotti, L.G. Immunobiology and pathogenesis of hepatitis B virus infection. Nat. Rev. Immunol. 2021.
- Yuen, M.F.; Chen, D.S.; Dusheiko, G.M.; Janssen, H.L.A.; Lau, D.T.Y.; Locarnini, S.A.; Peters, M.G.; Lai, C.L. Hepatitis B virus infection. Nat. Rev. Dis. Primers 2018, 4, 18035.
- Zoulim, F.; Locarnini, S. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology 2009, 137, 1593–1608.
- Buti, M.; Marcos-Fosch, C.; Esteban, R. Nucleos(t)ide analogue therapy: The role of tenofovir alafenamide. Liver Int. 2021, 41 (Suppl. 1), 9–14.
- Tu, T.; Budzinska, M.A.; Vondran, F.W.R.; Shackel, N.A.; Urban, S. Hepatitis B Virus DNA Integration Occurs Early in the Viral Life Cycle in an In Vitro Infection Model via Sodium Taurocholate Cotransporting Polypeptide-Dependent Uptake of Enveloped Virus Particles. J. Virol. 2018, 92.
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984.
- Rehermann, B.; Ferrari, C.; Pasquinelli, C.; Chisari, F.V. The hepatitis B virus persists for decades after patients’ recovery from acute viral hepatitis despite active maintenance of a cytotoxic T-lymphocyte response. Nat. Med. 1996, 2, 1104–1108.
- Revill, P.A.; Chisari, F.V.; Block, J.M.; Dandri, M.; Gehring, A.J.; Guo, H.; Hu, J.; Kramvis, A.; Lampertico, P.; Janssen, H.L.A.; et al. A global scientific strategy to cure hepatitis B. Lancet Gastroenterol. Hepatol. 2019, 4, 545–558.
- Jeng, W.J.; Lok, A.S.F. Is cure of hepatitis B infection a mission possible? In Hepatitis B Virus and Liver Disease; Kao, J.-H., Ed.; Springer: Singapore, 2021; pp. 475–495.
- Fanning, G.C.; Zoulim, F.; Hou, J.; Bertoletti, A. Therapeutic strategies for hepatitis B virus infection: Towards a cure. Nat. Rev. Drug Discov. 2019.
- Feng, S.; Gao, L.; Han, X.; Hu, T.; Hu, Y.; Liu, H.; Thomas, A.W.; Yan, Z.; Yang, S.; Young, J.A.T.; et al. Discovery of Small Molecule Therapeutics for Treatment of Chronic HBV Infection. ACS Infect. Dis. 2018, 4, 257–277.
- Prifti, G.M.; Moianos, D.; Giannakopoulou, E.; Pardali, V.; Tavis, J.E.; Zoidis, G. Recent Advances in Hepatitis B Treatment. Pharm. 2021, 14, 417.
- Ligat, G.; Verrier, E.R.; Nassal, M.; Baumert, T.F. Hepatitis B virus-host interactions and novel targets for viral cure. Curr. Opin. Virol. 2021, 49, 41–51.
- Bertoletti, A.; Le Bert, N. Immunotherapy for Chronic Hepatitis B Virus Infection. Gut Liver 2018, 12, 497–507.
- Lang, J.; Neumann-Haefelin, C.; Thimme, R. Immunological cure of HBV infection. Hepatol. Int. 2019, 13, 113–124.
- Gehring, A.J.; Protzer, U. Targeting Innate and Adaptive Immune Responses to Cure Chronic HBV Infection. Gastroenterology 2019, 156, 325–337.
- Schlicksup, C.J.; Zlotnick, A. Viral structural proteins as targets for antivirals. Curr. Opin. Virol. 2020, 45, 43–50.
- Kondylis, P.; Schlicksup, C.J.; Zlotnick, A.; Jacobson, S.C. Analytical Techniques to Characterize the Structure, Properties, and Assembly of Virus Capsids. Anal. Chem. 2019, 91, 622–636.
- Viswanathan, U.; Mani, N.; Hu, Z.; Ban, H.; Du, Y.; Hu, J.; Chang, J.; Guo, J.T. Targeting the multifunctional HBV core protein as a potential cure for chronic hepatitis B. Antivir. Res. 2020, 182, 104917.
- Martinez, M.G.; Boyd, A.; Combe, E.; Testoni, B.; Zoulim, F. Covalently closed circular DNA: The ultimate therapeutic target for curing HBV infections. J. Hepatol. 2021, 75, 706–717.
- Livingston, C.M.; Ramakrishnan, D.; Strubin, M.; Fletcher, S.P.; Beran, R.K. Identifying and Characterizing Interplay between Hepatitis B Virus X Protein and Smc5/6. Viruses 2017, 9, 69.
- Shen, C.; Feng, X.; Mao, T.; Yang, D.; Zou, J.; Zao, X.; Deng, Q.; Chen, X.; Lu, F. Yin-Yang 1 and HBx protein activate HBV transcription by mediating the spatial interaction of cccDNA minichromosome with cellular chromosome 19p13.11. Emerg. Microbes Infect. 2020, 9, 2455–2464.
- Tang, D.; Zhao, H.; Wu, Y.; Peng, B.; Gao, Z.; Sun, Y.; Duan, J.; Qi, Y.; Li, Y.; Zhou, Z.; et al. Transcriptionally inactive hepatitis B virus episome DNA preferentially resides in the vicinity of chromosome 19 in 3D host genome upon infection. Cell Rep. 2021, 35, 109288.
- Allweiss, L.; Giersch, K.; Pirosu, A.; Volz, T.; Muench, R.C.; Beran, R.K.; Urban, S.; Javanbakht, H.; Fletcher, S.P.; Lutgehetmann, M.; et al. Therapeutic shutdown of HBV transcripts promotes reappearance of the SMC5/6 complex and silencing of the viral genome in vivo. Gut 2021.
- Cheng, S.T.; Hu, J.L.; Ren, J.H.; Yu, H.B.; Zhong, S.; Wai Wong, V.K.; Kwan Law, B.Y.; Chen, W.X.; Xu, H.M.; Zhang, Z.Z.; et al. Dicoumarol, an NQO1 inhibitor, blocks cccDNA transcription by promoting degradation of HBx. J. Hepatol. 2021, 74, 522–534.
- Bock, C.T.; Schwinn, S.; Locarnini, S.; Fyfe, J.; Manns, M.P.; Trautwein, C.; Zentgraf, H. Structural organization of the hepatitis B virus minichromosome. J. Mol. Biol. 2001, 307, 183–196.
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979.
- Chabrolles, H.; Auclair, H.; Vegna, S.; Lahlali, T.; Pons, C.; Michelet, M.; Coute, Y.; Belmudes, L.; Chadeuf, G.; Kim, Y.; et al. Hepatitis B virus Core protein nuclear interactome identifies SRSF10 as a host RNA-binding protein restricting HBV RNA production. PLoS Pathog. 2020, 16, e1008593.
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 1221–1228.
- Lucifora, J.; Pastor, F.; Charles, E.; Pons, C.; Auclair, H.; Fusil, F.; Rivoire, M.; Cosset, F.L.; Durantel, D.; Salvetti, A. Evidence for long-term association of virion-delivered hepatitis B virus core protein with cccDNA independently of viral protein production. J. Hepatol. Rep. 2021.
- Qi, Y.; Gao, Z.; Xu, G.; Peng, B.; Liu, C.; Yan, H.; Yao, Q.; Sun, G.; Liu, Y.; Tang, D.; et al. DNA Polymerase kappa Is a Key Cellular Factor for the Formation of Covalently Closed Circular DNA of Hepatitis B Virus. PLoS Pathog. 2016, 12, e1005893.
- Tu, T.; Zehnder, B.; Qu, B.; Urban, S. De novo synthesis of hepatitis B virus nucleocapsids is dispensable for the maintenance and transcriptional regulation of cccDNA. JHEP Rep. 2021, 3, 100195.
- Zhang, Y.Y.; Zhang, B.H.; Theele, D.; Litwin, S.; Toll, E.; Summers, J. Single-cell analysis of covalently closed circular DNA copy numbers in a hepadnavirus-infected liver. Proc. Natl. Acad. Sci. USA 2003, 100, 12372–12377.
- Tuttleman, J.S.; Pourcel, C.; Summers, J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 1986, 47, 451–460.
- Makbul, C.; Nassal, M.; Böttcher, B. Slowly folding surface extension in the prototypic avian hepatitis B virus capsid governs stability. eLife 2020, 9.
- Ko, C.; Chakraborty, A.; Chou, W.M.; Hasreiter, J.; Wettengel, J.M.; Stadler, D.; Bester, R.; Asen, T.; Zhang, K.; Wisskirchen, K.; et al. Hepatitis B virus genome recycling and de novo secondary infection events maintain stable cccDNA levels. J. Hepatol. 2018, 69, 1231–1241.
- Revill, P.A.; Tu, T.; Netter, H.J.; Yuen, L.K.W.; Locarnini, S.A.; Littlejohn, M. The evolution and clinical impact of hepatitis B virus genome diversity. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 618–634.
- Nassal, M.; Junker-Niepmann, M.; Schaller, H. Translational inactivation of RNA function: Discrimination against a subset of genomic transcripts during HBV nucleocapsid assembly. Cell 1990, 63, 1357–1363.
- Bartenschlager, R.; Schaller, H. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 1992, 11, 3413–3420.
- Zhang, X.; Settembre, E.; Xu, C.; Dormitzer, P.R.; Bellamy, R.; Harrison, S.C.; Grigorieff, N. Near-atomic resolution using electron cryomicroscopy and single-particle reconstruction. Proc. Natl. Acad. Sci. USA 2008, 105, 1867–1872.
- Kühlbrandt, W. Biochemistry. The resolution revolution. Science 2014, 343, 1443–1444.
- Böttcher, B.; Nassal, M. Structure of Mutant Hepatitis B Core Protein Capsids with Premature Secretion Phenotype. J. Mol. Biol. 2018, 430, 4941–4954.
- Conley, M.J.; Bhella, D. Asymmetric analysis reveals novel virus capsid features. Biophys. Rev. 2019, 11, 603–609.
- Luque, D.; Caston, J.R. Cryo-electron microscopy for the study of virus assembly. Nat. Chem. Biol. 2020, 16, 231–239.
- Lecoq, L.; Fogeron, M.L.; Meier, B.H.; Nassal, M.; Bockmann, A. Solid-State NMR for Studying the Structure and Dynamics of Viral Assemblies. Viruses 2020, 12, 1069.
- Lutomski, C.A.; Lyktey, N.A.; Pierson, E.E.; Zhao, Z.; Zlotnick, A.; Jarrold, M.F. Multiple Pathways in Capsid Assembly. J. Am. Chem. Soc. 2018, 140, 5784–5790.
- Hadden, J.A.; Perilla, J.R.; Schlicksup, C.J.; Venkatakrishnan, B.; Zlotnick, A.; Schulten, K. All-atom molecular dynamics of the HBV capsid reveals insights into biological function and cryo-EM resolution limits. eLife 2018, 7.
- Perez-Segura, C.; Goh, B.C.; Hadden-Perilla, J.A. All-Atom MD Simulations of the HBV Capsid Complexed with AT130 Reveal Secondary and Tertiary Structural Changes and Mechanisms of Allostery. Viruses 2021, 13, 564.
- Patterson, A.; Zhao, Z.; Waymire, E.; Zlotnick, A.; Bothner, B. Dynamics of Hepatitis B Virus Capsid Protein Dimer Regulate Assembly through an Allosteric Network. ACS Chem. Biol. 2020, 15, 2273–2280.
- Makbul, C.; Khayenko, V.; Maric, H.M.; Bottcher, B. Conformational Plasticity of Hepatitis B Core Protein Spikes Promotes Peptide Binding Independent of the Secretion Phenotype. Microorganisms 2021, 9, 956.
- Zhao, Z.; Wang, J.C.; Segura, C.P.; Hadden-Perilla, J.A.; Zlotnick, A. The Integrity of the Intradimer Interface of the Hepatitis B Virus Capsid Protein Dimer Regulates Capsid Self-Assembly. ACS Chem. Biol. 2020, 15, 3124–3132.
- Perlmutter, J.D.; Hagan, M.F. Mechanisms of virus assembly. Annu. Rev. Phys. Chem. 2015, 66, 217–239.
- Zlotnick, A.; Johnson, J.M.; Wingfield, P.W.; Stahl, S.J.; Endres, D. A theoretical model successfully identifies features of hepatitis B virus capsid assembly. Biochemistry 1999, 38, 14644–14652.
- Wu, W.; Watts, N.R.; Cheng, N.; Huang, R.; Steven, A.C.; Wingfield, P.T. Expression of quasi-equivalence and capsid dimorphism in the Hepadnaviridae. PLoS Comput. Biol. 2020, 16, e1007782.
- Singh, S.; Zlotnick, A. Observed hysteresis of virus capsid disassembly is implicit in kinetic models of assembly. J. Biol. Chem. 2003, 278, 18249–18255.
- Chevreuil, M.; Lecoq, L.; Wang, S.; Gargowitsch, L.; Nhiri, N.; Jacquet, E.; Zinn, T.; Fieulaine, S.; Bressanelli, S.; Tresset, G. Nonsymmetrical Dynamics of the HBV Capsid Assembly and Disassembly Evidenced by Their Transient Species. J. Phys. Chem. B 2020, 124, 9987–9995.
- Lutomski, C.A.; Lyktey, N.A.; Zhao, Z.; Pierson, E.E.; Zlotnick, A.; Jarrold, M.F. Hepatitis B Virus Capsid Completion Occurs through Error Correction. J. Am. Chem. Soc. 2017, 139, 16932–16938.
- Spiriti, J.; Conway, J.F.; Zuckerman, D.M. Should Virus Capsids Assemble Perfectly? Theory and Observation of Defects. Biophys. J. 2020, 119, 1781–1790.
- Kondylis, P.; Schlicksup, C.J.; Katen, S.P.; Lee, L.S.; Zlotnick, A.; Jacobson, S.C. Evolution of Intermediates during Capsid Assembly of Hepatitis B Virus with Phenylpropenamide-Based Antivirals. ACS Infect. Dis. 2019, 5, 769–777.
- Zhang, M.; Zhang, J.; Tan, Y.; Xin, Y.; Gao, H.; Zheng, S.; Yi, Y.; Zhang, J.; Wu, C.; Zhao, Y.; et al. Efficacy and safety of GLS4/ritonavir combined with entecavir in HBeAg-positive patients with chronic hepatitis B: Interim results from phase 2b, multi-center study. J. Hepatol. 2020, 73, S878–S880.
- Feng, S.; Gane, E.; Schwabe, C.; Zhu, M.; Triyatni, M.; Zhou, J.; Bo, Q.; Jin, Y. A Five-in-One First-in-Human Study To Assess Safety, Tolerability, and Pharmacokinetics of RO7049389, an Inhibitor of Hepatitis B Virus Capsid Assembly, after Single and Multiple Ascending Doses in Healthy Participants. Antimicrob. Agents Chemother. 2020, 64.
- Yuen, M.F.; Zhou, X.; Gane, E.; Schwabe, C.; Tanwandee, T.; Feng, S.; Jin, Y.; Triyatni, M.; Lemenuel-Diot, A.; Cosson, V.; et al. Safety, pharmacokinetics, and antiviral activity of RO7049389, a core protein allosteric modulator, in patients with chronic hepatitis B virus infection: A multicentre, randomised, placebo-controlled, phase 1 trial. Lancet Gastroenterol. Hepatol. 2021, 6, 723–732.
- Berke, J.M.; Tan, Y.; Verbinnen, T.; Dehertogh, P.; Vergauwen, K.; Vos, A.; Lenz, O.; Pauwels, F. Antiviral profiling of the capsid assembly modulator BAY41-4109 on full-length HBV genotype A-H clinical isolates and core site-directed mutants in vitro. Antivir. Res. 2017, 144, 205–215.
- Zhou, Z.; Hu, T.; Zhou, X.; Wildum, S.; Garcia-Alcalde, F.; Xu, Z.; Wu, D.; Mao, Y.; Tian, X.; Zhou, Y.; et al. Heteroaryldihydropyrimidine (HAP) and Sulfamoylbenzamide (SBA) Inhibit Hepatitis B Virus Replication by Different Molecular Mechanisms. Sci. Rep. 2017, 7, 42374.
- Liu, Y.; Chang, S.; Hsieh, D.; Burdette, D.; Martin, R.; Mo, H.; Feierbach, B. Generation of an HBV core phenotyping assay for evaluating HBV capsid compounds. J. Virol. Methods 2021, 292, 114117.
- Luo, Y.; Cheng, J.; Hu, Z.; Ban, H.; Wu, S.; Hwang, N.; Kulp, J.; Li, Y.; Du, Y.; Chang, J.; et al. Identification of hepatitis B virus core protein residues critical for capsid assembly, pgRNA encapsidation and resistance to capsid assembly modulators. Antivir. Res. 2021, 191, 105080.
- Wu, S.; Luo, Y.; Viswanathan, U.; Kulp, J.; Cheng, J.; Hu, Z.; Xu, Q.; Zhou, Y.; Gong, G.Z.; Chang, J.; et al. CpAMs induce assembly of HBV capsids with altered electrophoresis mobility: Implications for mechanism of inhibiting pgRNA packaging. Antivir. Res. 2018, 159, 1–12.
- Lee, A.C.H.; Thi, E.P.; Ardzinski, A.; Brown, J.; Eley, T.; Mani, N.; Rijmbrand, R.; Sims, K.; Sofia, M.J.; Picchio, G. Hepatitis B virus core protein variants observed in a first-in-human placebo-controlled study of a core protein inhibitor. J. Hepatol. 2020, 73, S833.
- Verbinnen, T.; Hodari, M.; Talloen, W.; Berke, J.M.; Blue, D.; Yogaratnam, J.; Vandenbossche, J.; Shukla, U.; De Meyer, S.; Lenz, O. Virology analysis of chronic hepatitis B virus-infected patients treated for 28 days with JNJ-56136379 monotherapy. J. Viral Hepat. 2020, 27, 1127–1137.
- Verbinnen, T.; Talloen, W.; Shukla, U.; Vandenbossche, J.; Biermer, M.; Beumont-Mauviel, M.; De Meyer, S.; Lenz, O. Viral sequence analysis of chronic hepatitis B (CHB) patients treated with the capsid assembly modulator (CAM-N) JNJ-56136379 (JNJ-6379) as monotherapy in the ongoing JADE phase 2a study. (POSTER 856). Hepatology 2020, 72, 131A–1159A.
- Yuen, M.F.; Agarwal, K.; Gane, E.J.; Schwabe, C.; Ahn, S.H.; Kim, D.J.; Lim, Y.S.; Cheng, W.; Sievert, W.; Visvanathan, K.; et al. Safety, pharmacokinetics, and antiviral effects of ABI-H0731, a hepatitis B virus core inhibitor: A randomised, placebo-controlled phase 1 trial. Lancet Gastroenterol. Hepatol. 2020, 5, 152–166.
- Yuen, M.F.; Locarnini, S.; Revill, P.A.; Yan, R.; Ouyang, L.; Cai, D.; Delaney, W.; Kitrinos, K.M.; Thompson, A.; Zoulim, F.; et al. No emergent core inhibitor resistance in patients with chronic hepatitis B virus infection treated with Vebicorvir in combination with a nucleos (t)ide reverse transcriptase inhibitor. J. Hepatol. 2021, 75, S294–S803.