Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Allergy
Tryptophan is an essential amino acid whose metabolites play key roles in diverse physiological processes.They influence immune reactions, have antioxidant properties, and can function as anabolic signals. Apart from this, they act as regulators of the circadian rhythm, and can thus modulate sleep disorders or cognitive functions. Given these diverse roles, Trp metabolism has a significant impact on the clinical outcome of critically ill patients
- tryptophan metabolism
- kynurenine pathways
- sepsis
- inflammation
- sarcopenia
1. Introduction
Tryptophan (Trp) is the largest of the three aromatic amino acids, with a benzoic nucleus and a pyrrole ring on the side chain. With the molecular formula of C11H12N2O2, it is categorized as a glucogenic/ketogenic amino acid. It is also an essential amino acid since the human body lacks the enzymatic machinery for Trp synthesis. Of the eight essential amino acids, Trp has the lowest reserves in the body, and thus Trp deficiency rapidly manifests itself under various catabolic conditions. Hence, it is critical to consume Trp in sufficient quantities through the diet—rich sources include dairy products, eggs, meat, fish, and chocolate. Following its release from dietary protein, Trp passes through the intestinal epithelium into mesenteric and then into systemic circulation. Intestinal microbiota metabolizes Trp to indoles and thus delivers another important pool of tryptophan metabolites into systemic circulation.
Trp is metabolized through two pathways that include a series of active metabolic intermediates themselves capable of performing signaling functions (Figure 1).
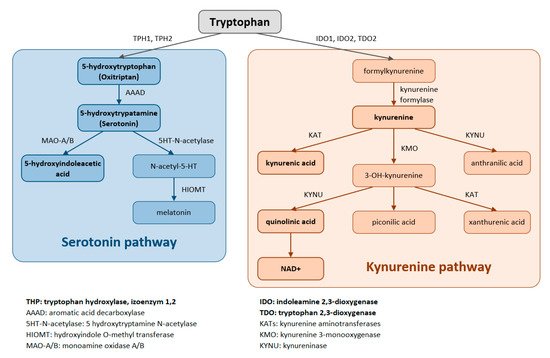
Figure 1. Tryptophan is metabolized by two pathways—the serotonin and the kynurenine pathways.
The aromatic Trp core is conserved in the serotonin pathway. The first step in the metabolic cascade is Trp hydroxylation to form 5-hydroxytryptophan (5-HTP, oxitriptan). This step is catalyzed by tryptophan hydroxylase (TPH), and of its two isoenzymes, TPH1 is active in peripheral tissues (gastrointestinal tract, skin) while TPH2 is active in neuronal cell types. Subsequently, aromatic acid decarboxylase (AAAD) removes the carboxyl group to form 5-hydroxytryptamine (5-HT, serotonin). The final product of this pathway is melatonin (N-acetyl-5-methoxy-tryptamine).
The majority (up to 90%) of all Trp is metabolized by the kynurenine pathway, where the intermediates are ligands for Aryl hydrocarbon receptors (AHRs) that primarily influence immune responses. In this metabolic pathway, the aromatic core is destroyed. The limiting step for its initiation is activation of two oxidoreductases—indolamine-2,3-dioxygenase (IDO) and tryptophan-2,3-dioxygenase (TDO). The IDO1 isoform is expressed in most tissues at low levels, including in macrophages, placenta, and epididymis, but not in the liver. The IDO2 isoform is found in the liver, brain, testes, endometrium, placenta, thyroid and can be induced in antigen- presenting cells and B lymphocytes. TDO is found mainly in the liver, but can also be expressed in bone marrow, the immune system, muscle tissue, the urinary and gastrointestinal tracts, and in the brain. Both IDO isoforms (IDO1, IDO2) are activated by proinflammatory cytokines, the most potent IDO1 activator being interferon γ. On the other hand, TDO is mainly activated by cortisol and the hypothalamic-pituitary-adrenal axis (HPA). The catabolites referred to as kynurenines include kynurenine (Kyn), kynurenic acid (KynA), quinolinic acid (QA) and the terminal products niacin (vitamin B3) and nicotinamide adenine dinucleotide (NAD+). The first rate-limiting step in the kynurenine pathway is conversion of Trp to N-formylkynurenine. This step is mediated by TDO in the liver. Under normal conditions, this is the major metabolic pathway. But expression levels of TDO are regulated by systemic levels of Trp, determined mainly by food intake and corticosteroids. Elevated levels of circulating glucocorticoids due to stress, sleep deprivation, or lack of physical activity, stimulate TDO expression. This leads to enhanced liver kynurenine metabolism and the subsequent increased delivery of kynurenines into the CNS. The situation is different with chronically high cortisol levels—it leads to glucocorticoid resistance, suppresses liver TDO expression and shifts to extrahepatic kynurenine metabolism.
IDO can be activated during inflammation (stimulated by TNF, interferon, etc.) to form Kyn, thus diminishing the amount of Trp available for the formation of serotonin, melatonin, and other important azoles (leading to impairment of sleep and cognitive function in critically ill patients). When the kynurenine pathway is fully activated (e.g., in sepsis with a typical increase in the Kyn/Trp ratio), it leads to Trp depletion, the consequent decreased activation of the rapamycin system and the attenuation of proteosynthesis. In critically ill patients, this might result in rapid progression of muscle atrophy, sarcopenia, and polyneuromyopathy [1,2,3].
2. The Role of Tryptophan in Sepsis and Inflammation
Inflammation is a vital response of the body to all forms of tissue damage caused variously by mechanical trauma, surgery, ischemic or thermal injury, or as a result of infection. The immune response initially remains local, but when there is severe tissue damage, it can degenerate into the so-called systemic inflammatory response syndrome (SIRS) or sepsis following an infection. The role of the inflammatory response is to restrict the area of damage, to stop the infection from spreading, and to repair damaged tissue and thus help restore tissue homeostasis. Since an out-of-control immune response can be lethal, its operation must be effectively coordinated. Sepsis, defined as life-threatening organ dysfunction caused by an unregulated host response to infection, is still one of the most common causes of death in critically ill patients [4]. “Metabolic immune regulation” modulates the immune response through local nutrient supply and Trp has a unique role to play in the modulation of this kind of homeostasis [5].
Immune cells recruited to an inflammatory lesion are exposed to many environmental stresses that trigger cellular signaling pathways. Similar to hypoxia, tissue inflammation activates hypoxia-inducible factor 1α, which modifies tissue metabolism to impaired oxygen supply by increasing the expression of genes responsible for glycolysis. Inflammation due to pro-inflammatory cytokines induces the expression of IDO—and thereby the Trp catabolic Kyn pathway—in antigen presenting cells (APCs: dendritic cells, macrophages, B lymphocytes), and in epithelial cells, vascular endothelial cells, and tumor cells. The simultaneous depletion of Trp and the production of Kyn modulates the immune response. IDO expression is tightly regulated by exogenous signals, and in the absence of inflammation plays only a minor role in Trp degradation. At the mRNA level, IDO transcription is promoted by forkhead box protein 3 (Foxo3) and interferon regulatory factor 8 (IRF-8). IDO is an intracellular enzyme and is not secreted, but although its metabolic effect is initially local, it is not only restricted to IDO-expressing cells. Both Trp deficiency and excess Kyn can be sensed by neighboring cells. As the tissue storage of Trp is low, it can be rapidly depleted by activated IDO, and patients with sepsis can thus be put at risk of inflammation [6] (Figure 2).
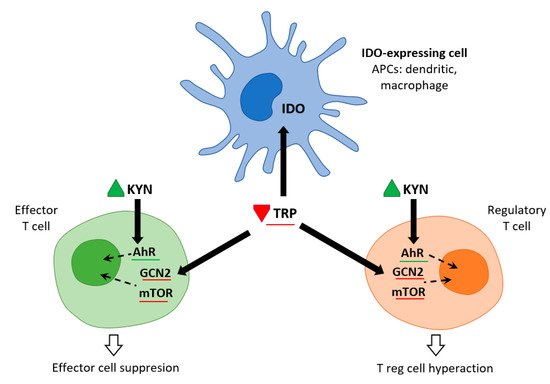
Figure 2. IDO and metabolic control of inflammation (adapted from [6]). IDO activity in APCs leads to Kyn release and Trp consumption. These metabolic signals affect both effector T cells and Treg cells via AhR (Kyn release) and the amino-acid sensors GCN2 and mTOR (Trp consumption), which in turn leads to immune suppression and tolerance.
Trp levels, which affect two important nutrient sensing systems, play a critical role in the metabolic regulation of inflammation. On the one hand, Trp activates general control non-depressible 2 stress kinase (GCN2) and on the other hand, it inhibits the mechanistic target of rapamycin complex1 (mTORC1). Depletion of Trp (either due to amino acid deficiency or imbalance) activates GCN2, leading to phosphorylation of its downstream target eukaryotic initiation factor 2α (eIF2α). In APCs, this is followed by the production of anti-inflammatory cytokines such as IL10 and the recruitment of regulatory T cells (Treg). Kynurenine can inhibit cytotoxicity of T cells and natural killer cells. This signaling role of IDO/GCN2 is not limited to immune cells but is involved in limiting inflammatory tissue damage, e.g., by inducing autophagy. Thus, activation of Trp metabolism has an anti-inflammatory and immunosuppressive effect, restoring homeostasis in the organism. On the other hand, Trp depletion blocks mTORC1 (a central regulator of cellular functions), which has a crucial role in proteosynthesis and in regulating skeletal muscle mass. The signaling role of IDO/mTORC1 is also important for immune cell activation [7,8].
3. Tryptophan, Kynurenine and Sarcopenia
Trp, like other amino acids, has a stimulatory effect on proteosynthesis. It acts directly by stimulating the rapamycin mTORC1 system. Its supplementation in the diet significantly increases IGF-1 in muscle and leads to the stimulation of proteosynthesis by activating the mTORC1/eIF4/p70s6k signaling pathway. In addition to the Trp signaling cascade directly inducing the expression of myogenic factors in C2 C12 myoblasts, a second Trp metabolic pathway is indirectly involved. Serotonin (5HT) stimulates growth hormone (GH) secretion from the pituitary gland, which increases IGF-1 as well as leptin production in the liver. Trp supplementation thus increases muscle mass and reduces adi-pose tissue. Trp is an amino acid commonly found as a component of total parenteral nutrition or amino acid solutions. The accumulation of Kyn has the opposite effect. The level of Trp decreases with age and this decrease, together with the accumulation of its catabolite Kyn, contributes to the development of sarcopenia [38,39].
Trp may be the key essential amino acid associated with sarcopenia. Metabolomic analyses show an association between low Trp and histidine levels and elevated glutamine and proline levels in age-related as well as cancer-related sarcopenia. As Trp is a glucogenic/ketogenic amino acid, a Trp deficient diet reduces glycolysis in muscle, and in turn increases the concentrations of amino acids, such as Pro and Gln, involved in the tricarboxylic acid cycle (TCA cycle). Glycolysis is linked to the mTORC1 pathway by glyceraldehyde-3-phosphate dehydrogenase binding to Rheb. All of this points to a unique role for Trp in the regulation of skeletal muscle mass, where its deficiency may suppress mTORC1 signaling via reduced glycolysis [31,40].
Exercise also activates LAT transporters, allowing more Trp and Kyn to enter the muscle fiber, while at the same time activating kynurenine aminotransferase (KAT), ensuring the transformation of Kyn to kynurenine acid. Muscle fiber contains only a fraction of the enzymatic equipment compared to liver or immune cells. Under normal conditions, 95% of Trp is metabolized by the Kyn pathway in the liver (TDO). TDO function is inhibited under conditions of stress, high levels of corticoids, inflammation, or sepsis, and Kyn production by IDO occurs mainly in immune cells (with feedback immunosuppression). Kynurenine enters the muscle fiber from circulating blood through LATs and is subsequently degraded to KynA by KAT, to 3Hydroxy-kynurenine (3HK), to quinolinic acid (QA) by kynurenine-3-monooxygenase (KMO), to anthranilic acid (AA) by kynureninase (KynU), and finally to nicotinamide adenine dinucleotide (NAD+) [37] (Figure 3).
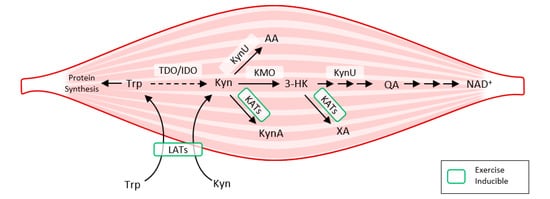
Figure 3. How exercise modulates tryptophan metabolism in skeletal muscle according to [38]. The kynurenine pathway of tryptophan degradation in skeletal muscle. Trp metabolites: Trp, Tryptophan; Kyn, Kynurenine; 3-HK, 3-Hydroxy-kynurenine; KynA, Kynurenic acid; AA, Anthranilic acid; QA, Quinolinic acid; Enzymes: TDO, Tryptophan 2,3-dioxygenase; IDO, Indoleamine 2,3-dioxygenase; KynU, Kynureninase; KATs, Kynurenine aminotransferases; KMO, Kynurenine 3-monooxygenase; LATs, large neutral amino acid transporters.
This entry is adapted from the peer-reviewed paper 10.3390/ijms222111714
This entry is offline, you can click here to edit this entry!