Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Microbiology
Tuberculosis (TB) is an infectious disease caused by Mycobacterium tuberculosis (Mtb), with 10.4 million new cases per year reported in the human population. Recent studies on the Mtb transcriptome have revealed the abundance of noncoding RNAs expressed at various phases of mycobacteria growth, in culture, in infected mammalian cells, and in patients. Among these noncoding RNAs are both small RNAs (sRNAs) between 50 and 350 nts in length and smaller RNAs (sncRNA) < 50 nts.
- mycobacteria
- small RNAs
- sncRNAs
- RNA processing
1. Introduction
Mycobacterium tuberculosis (Mtb) remains one of the leading infectious causes of human mortality, supplanted only in 2020 by the COVID-19 pandemic triggered by the SARS-CoV-2 virus. Mtb evolved from an ancestral smooth tubercule bacillus (e.g., M. canettii, M. pseudotuberculosis), acquiring virulence elements to attain its preferred pathogenicity towards humans [1]. The acquisition of these virulence elements coincided with Mtb undergoing a genomic downsizing relative to the 100 different smooth tubercule bacilli species characterized [1,2]. Despite this downsizing, a core genome is evident among the pathogenic strains of mycobacteria. Several decades of research efforts have been devoted to understanding how the ~4000 protein-coding elements evident in the Mtb genome contribute to growth, survival, and pathogenic processes [3,4,5,6,7]. Recent technical advances in deciphering the complex nature of Mtb and related mycobacterial genomes, including improved large-scale RNA-sequencing strategies, have revealed an abundance of small RNAs (sRNA). First, described as ranging in size from 50 to 350 nucleotides (nts) [8,9,10,11,12], these small RNAs now include some as small as 18 nts [13]. The sRNAs, originally selected with sequences > 100 nts in length, were found to represent ~11% of the intergenic transcripts (IGRs) identified from the exponential phase cultures. In addition to the sRNAs, IGRs include 5′ and 3′ UTRs, tRNAs, and antisense RNAs. Based on the normalized read counts for sense, antisense, and intergenic noncoding RNAs, the antisense and intergenic noncoding RNAs made up roughly 25% of the transcripts mapping outside of ribosomal RNA genes [10]. The sRNAs are detected in both exponential and/or stationary phase cultures, in infected eukaryotic cells, and in patients with tuberculosis (TB), suggesting key roles in all aspects of mycobacterial growth and survival [9,10,11,12,13,14].
2. Functional Roles of Mycobacterial sRNAs and sncRNAs
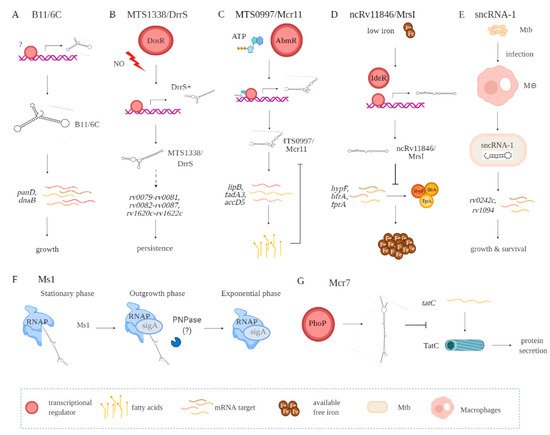
A key step in identifying putative biological roles for the sRNAs relates to what stages in a mycobacterial growth cycle they are expressed [41]. Additional insights have come from the environmental conditions that affect sRNA expression. Among the conditions are oxidative stress, nutrient deprivation, DNA damage, antibiotic exposure, and/or acidic environments, the latter occurring in the phagolysosome formed in macrophages and dendritic cells. Putative functional roles for the numerous sRNAs need also to consider the stability of the sRNA, affected by both the relative GC content and secondary RNA structures. Examples of several better characterized sRNAs are B11/6C, MTS1338/DrrS, Ms1, MTS0097/Mcr11, ncRv11846/MrsI, Mcr7, and sncRNA-1 (Figure 2, Table 1).
Figure 2. Functional contributions of seve ral Mtb-encoded sRNAs including B11/6C (A), MTS1338/DrrS (B), MTS0997/Mcr11 (C), ncRv11846/MrsI (D), sncRNA-1 (E), Ms1 (F), and Mcr7 (G). Indirect interactions are shown with a dashed line. (A) B11/6C is induced by undefined factors and positively regulates the expression genes (panD, dnaB) coupled to growth. (B) DrrS+ is induced by the DosR regulon upon nitric oxide stress. Then, it undergoes post transcriptional processing to yield MTS1338/DrrS. MTS1338/DrrS promotes the expression of three operons (rv0079-rv0081, rv0082-rv0087, and rv1620c-rv1622c), which cause defects in Mtb growth and promote persistence. The mechanism of this MTS1338/DrrS mediated regulation has not been characterized. (C) The expression of MTS0997/Mcr11 is regulated by AbmR, an ATP-bound transcription factor. After transcription, MTS0997/Mcr11 undergoes processing at the 3′ end and then regulates the expression of genes (lipB, fadA3, and accD5) involved in fatty acid production in a site-specific manner. This is negatively regulated by fatty acids. (D) In iron-restricted environments, the iron-responsive transcription factor IdeR induces the expression of ncRv11846/MrsI, which in return hinders the translation of nonessential iron storing proteins (hypF, bfrA, and fprA). This increases the level of free iron that can be used for essential functions. (E) sncRNA-1 is induced in infected macrophages and gets processed to yield 25 nts RNA. The processed sncRNA-1 enhances the expression of rv0242c and rv1094, two genes involved in oleic acid production. This regulatory network then promotes Mtb growth and survival inside macrophages. (F) Ms1 sequesters RNA polymerase (RNAP) at the stationary phase. Upon entrance to the outgrowth phase, Ms1 is degraded by PNPase, and some other RNases not yet identified, which release RNAP to promote global transcription. (G) PhoP induces the expression of Mcr7, which abrogates the translation of tatC. tatC encodes TatC, which is involved in the protein secretion pathway.
sRNA B11 (93 nts), later named 6C owing to its similarity to a small RNA found in other bacterial species, forms two stem-loops via six conserved cytosines [42,43]. Target sequence searches have suggested that B11/6C regulates Mtb transcripts coupled to DNA replication and protein secretion. In mechanistic studies in M. smegmatis, 6C was found to interact with two mRNA targets, panD and dnaB (Figure 2A, Table 1). Moreover, overexpression of 6C inhibited M. smegmatis growth. Several groups have used mycobacterial RNA over-expression vectors to further understand how the various sRNAs function. Over-expression of MTS1338 (117 nts) prevents Mtb replication, suggesting it targets key genes needed for mycobacterial growth (Figure 2B, Table 1) [19]. Later named as DosR regulated sRNA (DrrS), MTS1338 is induced by DosR. High levels of MTS2823 (300 nts) also inhibit Mtb growth, with transcriptome analysis using microarrays revealing that many transcripts involved in metabolism are downregulated (Figure 2B, Table 1) [10].
MTS0997 (131 nts), later named Mcr11, upregulates several genes required for Mtb fatty acid production (Figure 2C, Table 1) [30]. This sRNA positively regulates rv3282, fadA3, and lipB translation by binding a 7–11 nucleotide region upstream of the start codon. Supplementing fatty acids in the mycobacterial cultures overrides this regulatory process, revealing a feedback loop to control metabolic functions in Mtb. The regulatory role for sRNAs in Mtb metabolism is also revealed with ncRv11846 (106 nts). An ortholog of the E. coli sRNA RhyB, ncRv11846 is termed mycobacterial regulatory sRNA in iron (MrsI) (Figure 2D, Table 1) [12]. ncRv11846/MrsI is expressed following iron starvation [16]. This sRNA contains a six-nucleotide seed sequence that targets and negatively regulate the transcripts hypF and bfrA, which encode for nonessential iron-containing proteins. This translational roadblock increases the levels of free iron available. There are additional sRNAs identified that are reduced in expression in response to iron starvation [12]. The role of these sRNAs remains an open question.
Among the diverse sncRNAs, sncRNA-1 remains the best characterized (Figure 2E, Table 1) [13]. This non-coding RNA is present in the RD1 pathogenicity locus, in between esxA and espI. Over-expressing sncRNA-1 alters the Mtb transcriptome, with multiple genes required for fatty acid biogenesis increased in expression. Screening putative targets of sncRNA-1 by seed-sequence complementarity searches reveals two targets of this sRNA, rv0242c, and rv1094. These encode two proteins involved in the oleic acid biogenesis pathway. Both genes have putative sncRNA-1 binding sites within their 5′ UTRs. Substituting selected nucleotides involved in Watson‐Crick base pairing, either within the 5′ UTR or in the sncRNA seed sequence, eliminated the positive regulation. One novel approach for studying microRNA functions is the use of locked nucleic acid power inhibitors (LNA-PIs). These have modified RNA sequences that prevent their cleavage by RNA processing enzymes. They also have chemical modifications to enable uptake into cells without any transfection or liposome-based carrier needs [44]. They hybridize with target miRNAs with extremely high specificity, antagonizing the function of the miRNA. These LNA-PIs were tested in mycobacteria, which are inherently difficult to electroporate or transfect with liposome-based technologies. Notably, such LNAs are easily incorporated into mycobacteria and can antagonize sncRNAs in Mtb [13,45]. Incubation of Mtb with an LNA-PI selectively targeting sncRNA-1 abolished the upregulation of the rv0242c [13]. This LNA treatment reduced Mtb survival in infected macrophages, revealing a key pathogenic contribution of this sncRNA. The functions of sncRNA-6 and sncRNA-8 remain unexplored [13].
MTS2823 is termed Ms1 as it was functionally characterized in M. smegmatis and has homology to the 6S sRNA [26]. Best defined in E. coli, 6S sRNA has a secondary RNA structure that resembles an open promoter. The sigma factor bound RNA polymerase (RNAP) holoenzyme has a high affinity for this RNA structure [46]. The 6S sRNA complexes the RNAP, competitively reducing transcriptional activity [46]. Studies in M. smegmatis suggest that Ms1 competes with the sigma factor for binding to RNAP, hence suppressing transcriptional activity. Given the complexity of defining RNA‐protein complexes, a revised model is proposed in which Ms1 sequesters the RNAP (Figure 2F, Table 1) [27]. Another negative regulatory sRNA that has been characterized is Mcr7 [31]. This sRNA interferes with the translation of tatC mRNA, which encodes twin arginine translocation C (TatC) (Figure 2G, Table 1). TatC is a part of a protein export pathway that is also involved in Mtb pathogenesis [47]. All told, accumulating findings reveal a critical role for sRNAs and sncRNAs in Mtb pathogenicity.
3. Regulation of Mycobacterial sRNAs’/sncRNAs’ Expression
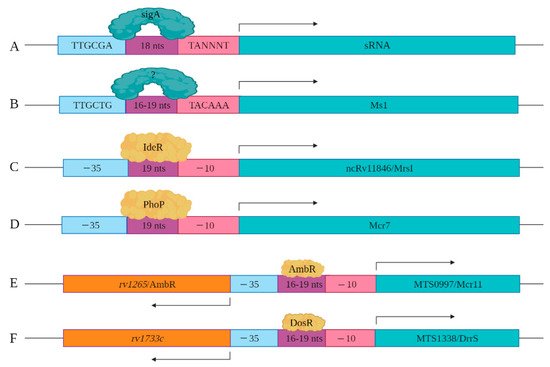
As more sRNAs/sncRNAs are discovered in mycobacteria, regulatory elements controlling their expression and processing are slowly being identified and characterized. This includes the identification of key cis- and trans-regulatory factors. In mycobacteria, sigA is the primary transcription factor, which is a member of the sigma70 family [48]. SigA recognizes the consensus cis regulatory sequence, the TTGCGA–N18–TANNNT hexamer that is present at −35 and −10 region upstream of the transcription start site (Figure 3A) [49,50]. SigA binding enables RNA polymerase to transcribe at promoter sites responsible for the expression of housekeeping regulons and for mycobacterial growth [48,51]. Miotto et al. developed computational predictions to identify sigA-regulated sRNAs [52]. Of the sRNAs identified in the screen, 46.9% had the consensus SigA promoter sequence in the upstream of the 5′ end, with 8.5% containing an intrinsic or factor-independent terminator sequence in the downstream or 3′ end. While 13.6% of the genes encoding sRNAs had both 5′ and 3′ motifs, their presence and impact on transcription requires further study. The remaining 31.0% of the sRNA encoding genes had neither defined motif, suggesting the involvement of other regulatory factors. For example, the gene encoding Ms1 contains a −10 element, starting five-nucleotide upstream of +1 position along with a distinct −35 element, suggesting that a distinct sigma factor regulates its expression (Figure 3B). Ms1 contains different regulatory elements (−491/+9 region) that contribute to its expression [27].
Figure 3. Cis and trans regulatory elements involved in sRNA expression are shown. (A) sigA recognizes a consensus sequence to induce the expression of a set of sRNAs identified by Miotto et al. (B) −35 and −10 elements upstream of Ms1 are shown. Distal regulatory elements not shown here also contribute to the expression of Ms1. (C) IdeR potentially regulates the expression of ncRv11846/MrsI through the IdeR-box found in its promoter region. (D) The expression of Mcr7 is regulated by PhoP, which is a part of the PhoPR two-component system. (E) The expression of MTS0997/Mcr11 is regulated by AmbR located in the upstream of MTS0997/Mcr11 and expressed in the opposite orientation. (F) The expression of MTS1338/DrrS is regulated by the DosR transcription factor.
Coupled with the cis-regulatory elements, novel trans-regulatory elements are being identified that control sRNAs/sncRNA expression. Among these are alternate transcription factors or sigma factors. For instance, sRNA ncRv11846/MrsI has an IdeR binding site in its promoter region. IdeR is an iron-responsive master regulator of genes coupled to iron metabolism, including the sRNA ncRv11846/MrsI (Figure 3C) [12,53]. Mcr7 expression is regulated by PhoP, which is a part of the two-component system PhoP/PhoR [31,54]. Direct binding assays with chromatin immuno-precipitation of PhoP revealed that it binds to the promoter region of Mcr7 to induce its expression in exponential phase Mtb cultures (Figure 3D). sRNA MTS0997/Mcr11 resides between two protein-coding genes rv1264 and rv1265, with the protein products of these two genes involved in the metabolism of cAMP [14]. rv1264 encodes an adenylyl cyclase, which catalyzes ATP to cAMP. rv1265 is a transcription factor that binds to both ATP and DNA. DNA binding studies have shown that rv1265 induces MTS0997/Mcr11 expression (Figure 3E). rv1265 is now termed AbmR for ATP binding Mcr11 regulator. Mapping studies of the 5′ end of MTS0997/Mcr11 revealed that its −35 element coincides with the promoter regions of AmbR, which is oriented in the opposite direction (Figure 3E) [10]. MTS1338/DrrS is also transcribed in the opposite direction to its neighboring gene called rv1733c, but mapping of the TSS of rv1733c revealed that it is separated by 190 nucleotides from the TSS of MTS1338/DrrS [55]. rv1733c encodes a protein involved in cell wall biogenesis and is a component of the DosR regulon [10]. The DosR regulon, induced by nitric oxide (NO), is the primary mediator of the hypoxic stress response [56]. MTS1338/DrrS is also upregulated in response to NO, and the MTS1338/DrrS promoter is activated by DosR, established with b-galactosidase reporter assays (Figure 3F) [28].
In summary, identification of the cis- and trans-acting factors is revealing many diverse types or regulatory elements involved in the sRNA expression. Little is known about the regulation of the sncRNAs.
4. Processing of Mycobacterial sRNAs and sncRNAs
Many sRNAs are generated as full-length mature transcripts with no obvious processing steps. Yet, several of the smaller species do undergo some form of processing from larger single-stranded (ssRNA) precursors [27,28,30]. Among these are Ms1, MTS0997/Mcr11, MTS1338/DrrS, sncRNA-1, and sncRNA-6. Ms1 is a 300 nt transcript detected in both exponential and stationary phase cultures. Notably, it also exists as a 250 nt transcript in stationary phase, suggesting some form of processing [10]. MTS1338/DrrS is transcribed as a precursor transcript of >400 nts (referred to as DrrS+) that is cleaved at the 3′ end to yield the mature 108 nts form [28]. MTS0997/Mcr11 has a 3′ end that varies in size by 3–14 nts, implying that a 3′ RNA processing occurs like that for MTS133/DrrS [30].
Both sncRNA-1 and sncRNA-6, which have final sizes of 25 nts and 21 nts, respectively, require processing enzymes for their generation [13]. These sncRNAs were predicted to exist as precursor transcripts >115 nts that have defined RNA structures involving double-stranded RNAs (dsRNA) segments that form hairpin loops. To identify the putative processing requirements needed for the generation of sncRNA-1, nucleotide substitutions were created within the hairpin loop and antisense complementarity strand of the precursor form of this sncRNA. This caused the formation of multiple intermediate size-transcripts (40–115 nts), detected by Northern blotting [13]. Thus, the processing of the longer RNA transcript depends on both the formation of the hairpin loop and the specific nucleotides at a putative cleavage site needed to form sncRNA-1 [13]. Notably, the expression of the precursor sncRNA-1 transcript, containing sncRNA-1 that was no longer processed into the 25 nt species because of the introduction of nucleotide substitutions, was unable to regulate gene expression. SncRNA-6 also undergoes a sequence-specific processing from a longer RNA transcript. Like sncRNA-1, mutations that disrupt the hairpin loop in which sncRNA-6 resides or the mutations at the cleavage site of sncRNA-6 prevent its processing. Taken together, multiple experiments establish the existence of a small RNA processing system in mycobacteria. These findings do not exclude the possibility that some of the Mtb sRNAs could be generated by miRNA processing enzymes when the mycobacteria are propagating in eukaryotic cells during infections [57].
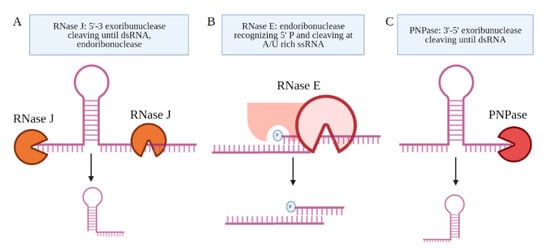
Several candidate RNA processing enzymes have been reported to date. Among these are ribonuclease E (RNase E), polynucleotide phosphorylase (PNPase or GpsI), ribonuclease J (RNase J), and the ATP-dependent RNA helicase RhlE (Figure 4) [58]. All are components of the RNA degradosome. Except RhlE, all are essential for in vitro growth, determined by identifying key genes through a transposon mutagenesis screen (Himar1 transposon libraries) [7]. Mechanistically, RNase E recognizes the 5′ phosphate of the transcript and then cuts at an A/U rich sequence of the ssRNA [59]. PNPase and RNase J are 3′ and 5′ specific exonucleases, respectively, that stop upon the presence of a dsRNA sequence [60]. Many research teams have made use of CRISPR interference mediated knock down of the RNA processing enzymes to study their role in the generation of specific sRNAs [27,58,61]. Sikova et al. has investigated the contribution of the core RNase enzymes in the processing of Ms1 [27]. Knockdown of PNPase increased the levels of Ms1 ~30%, while the targeting of RNase E and RNase J had no effect on this sRNA, revealing some target specificity. These findings further suggest that the processing of Ms1 likely involves additional RNA processing enzymes. Another possibility is that residual protein levels of PNPase were still resulting in some processing of the longer RNA transcript. Taken together, the limited number of studies on the RNA processing enzymes leave open many questions about how Mtb produces sRNAs from longer transcripts.
Figure 4. The proteins coupled to the Mtb degradosome are shown. (A) RNase J is an endoribonuclease and a 5′–3′ exoribonuclease. (B) RNase E is an endoribonuclease that cleaves ssRNA at A/U-rich sites after recognizing the 5′ phosphate in proximity. (C) PNPase is a 3′–5′ exoribonuclease, which is the only RNase implicated in the processing of an sRNA, Ms1.
This entry is adapted from the peer-reviewed paper 10.3390/ncrna7040069
This entry is offline, you can click here to edit this entry!