Cardiotoxicity is a well-recognized late effect among childhood cancer survivors. With various pediatric cancers becoming increasingly curable, it is imperative to understand the disease burdens that survivors may face in the future.
- cardio-oncology
- pediatric cancer
- cardiotoxicity
- anthracyclines
- pediatric heart failure
1. Introduction
Heart failure can be an early or late cardiotoxic side effect of cancer treatment in children. Anthracycline cardiotoxicity is characterized as acute if it occurs within the first week of treatment, early-onset progressive cardiotoxicity if it occurs within the first year of treatment completion, and late-onset progressive cardiotoxicity if it occurs more than one year after the completion of treatment [1].
2. Pathophysiology of Cardiotoxic Cancer Therapies
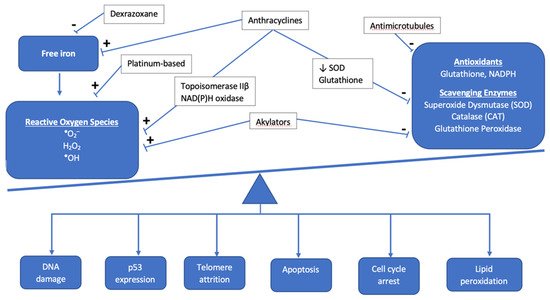
2.1. Anthracyclines
| Genetic Modifiers of Cardiotoxicity Risk | |
|---|---|
| Deleterious Effect | |
| Gene/SNP | Mechanism of toxicity |
| RARG (S427L variant) | Increase topoisomerase IIβ expression |
| rs1883112 SNP, p40phox subunit NAD(P)H oxidase | Interstitial fibrosis |
| UGT1A6*A | Decreased drug glucuronidation and clearance |
| MYH7 | Variants documented in familial dilated cardiomyopathy Downregulated in hiPSC-CM treated with doxorubicin |
| Titin truncating variants (TTN) | Encode A and I bands in sarcomeres; associated with depressed LV function |
| Indeterminant Effect | |
| rs4673 SNP, p22phos subunit NAD(P)H | Protective against myocardial fibrosis Acute cardiotoxicity |
2.2. Non-Anthracycline Agents
2.3. Radiation-Induced Cardiotoxicity
2.3.1. Mechanisms for Toxicity
2.3.2. Dose-Toxicity Relationship
| First Author | n | Treatment Period | Age at Treatment, Years | Length of Follow-Up, Years (Median) | Risk of Grade ≥ 3 Cardiotoxicity (95% CI) |
|---|---|---|---|---|---|
| Haddy [37] | 3162 | 1942–1985 | <17 | 26 | No anthracycline:
|
| van der Pal [38] | 1362 | 1966–1996 | <18 | 22 | Radiation only: HR 13.0 (2.8–61) Anthracycline & radiation: HR 49.5 (10.7–230) HR 1.8 (1.4–2.2) per 10 Gy to heart |
| Mulrooney [30] | 23,462 | 1970–1999 | <20 (median 6) | 28 | HF:
|
2.4. Targeted Cancer Therapies
2.5. Cellular Therapy and Hematopoietic Stem Cell Transplantation
This entry is adapted from the peer-reviewed paper 10.3390/children8090829
References
- Lipshultz, S.E.; Adams, M.J.; Colan, S.D.; Constine, L.S.; Herman, E.H.; Hsu, D.T.; Hudson, M.M.; Kremer, L.C.; Landy, D.C.; Miller, T.L.; et al. Long-term cardiovascular toxicity in children, adolescents, and young adults who receive cancer therapy: Pathophysiology, course, monitoring, management, prevention, and research directions: A scientific statement from the American Heart Association. Circulation 2013, 128, 1927–1995.
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879.
- Mulrooney, D.A.; Yeazel, M.W.; Kawashima, T.; Mertens, A.C.; Mitby, P.; Stovall, M.; Donaldson, S.S.; Green, D.M.; Sklar, C.A.; Robison, L.L.; et al. Cardiac outcomes in a cohort of adult survivors of childhood and adolescent cancer: Retrospective analysis of the Childhood Cancer Survivor Study cohort. BMJ 2009, 339, b4606.
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 2012, 137289.
- Deidda, M.; Madonna, R.; Mango, R.; Pagliaro, P.; Bassareo, P.P.; Cugusi, L.; Romano, S.; Penco, M.; Romeo, F.; Mercuro, G. Novel insights in pathophysiology of antiblastic drugs-induced cardiotoxicity and cardioprotection. J. Cardiovasc. Med. 2016, 17 (Suppl. 1), S76–S83.
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642.
- Goormaghtigh, E.; Chatelain, P.; Caspers, J.; Ruysschaert, J.M. Evidence of a complex between adriamycin derivatives and cardiolipin: Possible role in cardiotoxicity. Biochem. Pharmacol. 1980, 29, 3003–3010.
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250.
- Qiao, X.; van der Zanden, S.Y.; Wander, D.P.A.; Borràs, D.M.; Song, J.Y.; Li, X.; van Duikeren, S.; van Gils, N.; Rutten, A.; van Herwaarden, T.; et al. Uncoupling DNA damage from chromatin damage to detoxify doxorubicin. Proc. Natl. Acad. Sci. USA 2020, 117, 15182–15192.
- Abramson, J.J.; Buck, E.; Salama, G.; Casida, J.E.; Pessah, I.N. Mechanism of anthraquinone-induced calcium release from skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 1988, 263, 18750–18758.
- Sag, C.M.; Köhler, A.C.; Anderson, M.E.; Backs, J.; Maier, L.S. CaMKII-dependent SR Ca leak contributes to doxorubicin-induced impaired Ca handling in isolated cardiac myocytes. J. Mol. Cell. Cardiol. 2011, 51, 749–759.
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630.
- Keltai, K.; Cervenak, L.; Makó, V.; Doleschall, Z.; Zsáry, A.; Karádi, I. Doxorubicin selectively suppresses mRNA expression and production of endothelin-1 in endothelial cells. Vascul. Pharmacol. 2010, 53, 209–214.
- Magdy, T.; Burmeister, B.T.; Burridge, P.W. Validating the pharmacogenomics of chemotherapy-induced cardiotoxicity: What is missing? Pharmacol. Ther. 2016, 168, 113–125.
- Aminkeng, F.; Bhavsar, A.P.; Visscher, H.; Rassekh, S.R.; Li, Y.; Lee, J.W.; Brunham, L.R.; Caron, H.N.; van Dalen, E.C.; Kremer, L.C.; et al. A coding variant in RARG confers susceptibility to anthracyclineinduced cardiotoxicity in childhood cancer. Nat. Genet. 2015, 47, 1079–1084.
- Wojnowski, L.; Kulle, B.; Schirmer, M.; Schluter, G.; Schmidt, A.; Rosenberger, A.; Vonhof, S.; Bickeboller, H.; Toliat, M.R.; Suk, E.K.; et al. NAD(P)H oxidase and multidrug resistance protein genetic polymorphisms are associated with doxorubicin-induced cardiotoxicity. Circulation 2005, 112, 3754–3762.
- Rossi, D.; Rasi, S.; Franceschetti, S.; Capello, D.; Castelli, A.; De Paoli, L.; Ramponi, A.; Chiappella, A.; Pogliani, E.M.; Vitolo, U.; et al. Analysis of the host pharmacogenetic background for prediction of outcome and toxicity in diffuse large B-cell lymphoma treated with R-CHOP21. Leukemia 2009, 23, 1118–1126.
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23.
- Cascales, A.; Pastor-Quirante, F.; Sanchez-Vega, B.; Luengo-Gil, G.; Corral, J.; Ortuno-Pacheco, G.; Vicente, V.; de la Pena, F.A. Association of anthracycline-related cardiac histological lesions with NADPH oxidase functional polymorphisms. Oncologist 2013, 18, 446–453.
- Garcia-Pavia, P.; Kim, Y.; Restrepo-Cordoba, M.A.; Lunde, I.G.; Wakimoto, H.; Smith, A.M.; Toepfer, C.N.; Getz, K.; Gorham, J.; Patel, P.; et al. Genetic Variants Associated With Cancer Therapy-Induced Cardiomyopathy. Circulation 2019, 140, 31–41.
- Zhang, X.; Zhu, Y.; Dong, S.; Zhang, A.; Lu, Y.; Li, Y.; Lv, S.; Zhang, J. Role of oxidative stress in cardiotoxicity of antineoplastic drugs. Life Sci. 2019, 232, 116526.
- Rtibi, K.; Grami, D.; Selmi, S.; Amri, M.; Sebai, H.; Marzouki, L. Vinblastine, an anticancer drug, causes constipation and oxidative stress as well as others disruptions in intestinal tract in rat. Toxicol. Rep. 2017, 4, 221–225.
- Madeddu, C.; Deidda, M.; Piras, A.; Cadeddu, C.; Demurtas, L.; Puzzoni, M.; Piscopo, G.; Scartozzi, M.; Mercuro, G. Pathophysiology of cardiotoxicity induced by nonanthracycline chemotherapy. J. Cardiovasc. Med. 2016, 17 (Suppl. 1), e12–e18.
- Tocchetti, C.G.; Cadeddu, C.; Di Lisi, D.; Femminò, S.; Madonna, R.; Mele, D.; Monte, I.; Novo, G.; Penna, C.; Pepe, A.; et al. From Molecular Mechanisms to Clinical Management of Antineoplastic Drug-Induced Cardiovascular Toxicity: A Translational Overview. Antioxid Redox Signal. 2019, 30, 2110–2153.
- Lamberti, M.; Porto, S.; Zappavigna, S.; Addeo, E.; Marra, M.; Miraglia, N.; Sannolo, N.; Vanacore, D.; Stiuso, P.; Caraglia, M. A mechanistic study on the cardiotoxicity of 5-fluorouracil in vitro and clinical and occupational perspectives. Toxicol. Lett. 2014, 227, 151–156.
- Koutroumpakis, E.; Palaskas, N.L.; Lin, S.H.; Abe, J.I.; Liao, Z.; Banchs, J.; Deswal, A.; Yusuf, S.W. Modern Radiotherapy and Risk of Cardiotoxicity. Chemotherapy 2020, 65, 65–76.
- Nielsen, K.M.; Offersen, B.V.; Nielsen, H.M.; Vaage-Nilsen, M.; Yusuf, S.W. Short and long term radiation induced cardiovascular disease in patients with cancer. Clin. Cardiol. 2017, 40, 255–261.
- Aleman, B.M.; van den Belt-Dusebout, A.W.; De Bruin, M.L.; van ’t Veer, M.B.; Baaijens, M.H.; de Boer, J.P.; Hart, A.A.; Klokman, W.J.; Kuenen, M.A.; Ouwens, G.M.; et al. Late cardiotoxicity after treatment for Hodgkin lymphoma. Blood 2007, 109, 1878–1886.
- van Nimwegen, F.A.; Schaapveld, M.; Janus, C.P.; Krol, A.D.; Petersen, E.J.; Raemaekers, J.M.; Kok, W.E.; Aleman, B.M.; van Leeuwen, F.E. Cardiovascular disease after Hodgkin lymphoma treatment: 40-year disease risk. JAMA Intern. Med. 2015, 175, 1007–1017.
- Mulrooney, D.A.; Hyun, G.; Ness, K.K.; Ehrhardt, M.J.; Yasui, Y.; Duprez, D.; Howell, R.M.; Leisenring, W.M.; Constine, L.S.; Tonorezos, E.; et al. Major cardiac events for adult survivors of childhood cancer diagnosed between 1970 and 1999: Report from the Childhood Cancer Survivor Study cohort. BMJ 2020, 368, l6794.
- Cutter, D.J.; Schaapveld, M.; Darby, S.C.; Hauptmann, M.; van Nimwegen, F.A.; Krol, A.D.; Janus, C.P.; van Leeuwen, F.E.; Aleman, B.M. Risk of valvular heart disease after treatment for Hodgkin lymphoma. J. Natl. Cancer Inst. 2015, 107.
- Hancock, S.L.; Tucker, M.A.; Hoppe, R.T. Factors affecting late mortality from heart disease after treatment of Hodgkin’s disease. JAMA 1993, 270, 1949–1955.
- van Nimwegen, F.A.; Schaapveld, M.; Cutter, D.J.; Janus, C.P.; Krol, A.D.; Hauptmann, M.; Kooijman, K.; Roesink, J.; van der Maazen, R.; Darby, S.C.; et al. Radiation Dose-Response Relationship for Risk of Coronary Heart Disease in Survivors of Hodgkin Lymphoma. J. Clin. Oncol. 2016, 34, 235–243.
- Armenian, S.H.; Hudson, M.M.; Mulder, R.L.; Chen, M.H.; Constine, L.S.; Dwyer, M.; Nathan, P.C.; Tissing, W.J.; Shankar, S.; Sieswerda, E.; et al. Recommendations for cardiomyopathy surveillance for survivors of childhood cancer: A report from the International Late Effects of Childhood Cancer Guideline Harmonization Group. Lancet Oncol. 2015, 16, e123–e136.
- van Nimwegen, F.A.; Ntentas, G.; Darby, S.C.; Schaapveld, M.; Hauptmann, M.; Lugtenburg, P.J.; Janus, C.P.M.; Daniels, L.; van Leeuwen, F.E.; Cutter, D.J.; et al. Risk of heart failure in survivors of Hodgkin lymphoma: Effects of cardiac exposure to radiation and anthracyclines. Blood 2017, 129, 2257–2265.
- Martel, M.K.; Sahijdak, W.M.; Ten Haken, R.K.; Kessler, M.L.; Turrisi, A.T. Fraction size and dose parameters related to the incidence of pericardial effusions. Int. J. Radiat. Oncol. Biol. Phys. 1998, 40, 155–161.
- Haddy, N.; Diallo, S.; El-Fayech, C.; Schwartz, B.; Pein, F.; Hawkins, M.; Veres, C.; Oberlin, O.; Guibout, C.; Pacquement, H.; et al. Cardiac Diseases Following Childhood Cancer Treatment: Cohort Study. Circulation 2016, 133, 31–38.
- van der Pal, H.J.; van Dalen, E.C.; van Delden, E.; van Dijk, I.W.; Kok, W.E.; Geskus, R.B.; Sieswerda, E.; Oldenburger, F.; Koning, C.C.; van Leeuwen, F.E.; et al. High risk of symptomatic cardiac events in childhood cancer survivors. J. Clin. Oncol. 2012, 30, 1429–1437.
- Ganatra, S.; Parikh, R.; Neilan, T.G. Cardiotoxicity of Immune Therapy. Cardiol. Clin. 2019, 37, 385–397.
- Rotz, S.J.; Ryan, T.D.; Hlavaty, J.; George, S.A.; El-Bietar, J.; Dandoy, C.E. Cardiotoxicity and cardiomyopathy in children and young adult survivors of hematopoietic stem cell transplant. Pediatr. Blood Cancer 2017, 64, e26600.
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73.
- Ganatra, S.; Carver, J.R.; Hayek, S.S.; Ky, B.; Leja, M.J.; Lenihan, D.J.; Lenneman, C.; Mousavi, N.; Park, J.H.; Perales, M.A.; et al. Chimeric Antigen Receptor T-Cell Therapy for Cancer and Heart: JACC Council Perspectives. J. Am. Coll. Cardiol. 2019, 74, 3153–3163.
- Le, R.Q.; Li, L.; Yuan, W.; Shord, S.S.; Nie, L.; Habtemariam, B.A.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Tocilizumab for Treatment of Chimeric Antigen Receptor T Cell-Induced Severe or Life-Threatening Cytokine Release Syndrome. Oncologist 2018, 23, 943–947.
- Pathan, N.; Hemingway, C.A.; Alizadeh, A.A.; Stephens, A.C.; Boldrick, J.C.; Oragui, E.E.; McCabe, C.; Welch, S.B.; Whitney, A.; O’Gara, P.; et al. Role of interleukin 6 in myocardial dysfunction of meningococcal septic shock. Lancet 2004, 363, 203–209.