The potential of computational models to identify new therapeutics and repurpose existing drugs has gained significance in recent times. The current ‘COVID-19’ pandemic caused by the new SARS CoV2 virus has affected over 200 million people and caused over 4 million deaths. The enormity and the consequences of this viral infection have fueled the research community to identify drugs or vaccines through a relatively expeditious process. The availability of high-throughput datasets has cultivated new strategies for drug development and can provide the foundation towards effective therapy options. Molecular modeling methods using structure-based or computer-aided virtual screening can potentially be employed as research guides to identify novel antiviral agents.
1. Introduction
The identification of effective therapeutics for COVID-19 has been an important focus of research across the world ever since its emergence. Interestingly, with two years of effort in this domain, many approaches have been taken to recognize and develop potential candidates against this virus, with some being more efficient than others. This fast-paced pursuit for the identification of therapeutics has brought in-silico and computational modeling into the limelight, mainly driven by the inefficiency of experimental high-throughput screening (HTS) for COVID-19 drug discovery [
1]. Computational modeling holds the key to this bottleneck by providing a more efficient solution to expedite drug discovery or repurposing for the COVID-19 pandemic by evaluating the biological interactions of drugs with viral molecular components based on existing datasets [
2,
3,
4]. There are two types of computational approaches towards identifying therapeutics: target-based and disease-based. The former generates information on the drug–target interaction, while the latter utilizes existing databases to determine new indications of existing drugs via comparisons with disease characteristics [
5,
6].
Another popular mode of drug discovery is virtual screening (VS), an approach that combines initial computing followed by experimental targeting of specific biomolecules, such as DNA, protein, RNA, or lipid molecules, for the purpose of inhibiting or activating them [
7]. This facilitates the rapid identification of viral proteins, with their structures determined using experimental and computational methods. Unlike conventional vaccine discovery, this approach provides a cost- and time-efficient route towards the identification of potential drug candidates for several microbial pathogens. Reverse vaccinology (RV), an approach that uses the genome to design vaccines, has revolutionized the field, as bacteria no longer need to be cultured to identify vaccine targets [
8]. As a consequence, all putative target protein antigens can be identified, instead of being restricted to those isolated from bacterial cultures. Considering these benefits, researchers are focusing on developing RV prediction algorithms. Currently, there are no licensed drugs suitable for such viral infections. Consequently, drug repurposing has become the best course of action in this scenario, involving the effective adaptation of existing antiviral agents for alternative treatments. Several FDA-approved drug candidates that have been previously applied for SARS CoV1 and Middle Eastern Respiratory Syndrome (MERS) management are currently being investigated for SARS CoV2 therapy. Drug repurposing plays a specific role in drug discovery by identifying new therapeutic uses for already studied drugs [
9,
10,
11]. Globally, more than 500 clinical trials of repurposed drugs have been registered and a further 150 have been initiated [
5]. The current research studies are mainly focused on the utilization of antiviral drugs and repurposing strategies, as well as vaccine development [
6,
12,
13,
14,
15,
16,
17]. The WHO roadmap [
18] provides the necessary guidelines and support for regulation, ethics, and platform utilization to ensure the efficient development of vaccines and treatments.
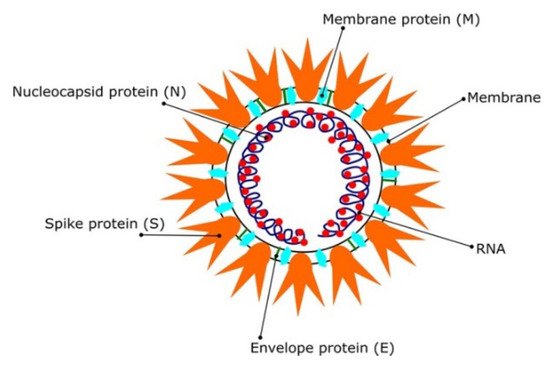
Most coronaviruses share the same genetic features. As a result of their large genome size (about 27–32 kilobases), they are classified as RNA viruses with positive sense. Because they have overlapping ORF1a and ORF1b polyproteins, their genomes are not segmented. Polyproteins, as shown in
Figure 1, arecomposed of 16 non-structural proteins (NSPs) and structural proteins in the form of spike (S), envelope (E), membrane (M), and nucleoproteins (N) [
19]. After infection of host cells, different CoVs have been shown to undergo genome recombination [
20]. This recombination contributes to their evolution into new types, which could include new intermediate hosts. As a result, CoVs have strong adaptation ability and the ability to be involved in a wide range of species; however, the identification of these viruses’ spike glycoproteins is crucial for vaccine development. In addition, the use of in-silico studies to design new antiviral drug candidates is beneficial [
21,
22]. This is why the targeting of SARS CoV2 spike (S) proteins, polyproteins, or other viral proteins for vaccine and therapeutic development is becoming increasingly essential approach [
23].
Figure 1. The SARS CoV2 virions structure. Schematic representation reproduced with permission from reference [
12]. Copyright © 2021 Informa UK Limited, trading as Taylor & Francis Group.
Alternatively, natural plant-based bioactive compounds have been proven to possess antiviral properties that combat infection at various stages of the viral replication cycle, including virus entry inhibition into the host cells [
24,
25,
26,
27]. In addition to therapeutic identification, delivery is a crucial part of drug discovery. Nano-carriers hold huge potential in drug delivery and can be easily adapted for vaccine development or antiviral delivery targeting specific peptides of CoV protein [
28,
29,
30]. Many datasets on the structure–activity relationships and toxicity of nano delivery systems are available as a consequence of the booming popularity of this field, which can be adapted for the current pandemic as well [
31,
32,
33,
34].
2. In Silico Modeling in Vaccine Development: A SARS CoV-2 Case Study
While antiviral identification and development help in managing viral infections, vaccine development will offer herd immunity in the longer run [
73,
74,
75]. Currently, there are more than 20 vaccines being tested in clinical trials. The World Health Organization (WHO) is regularly updating its list of the developed vaccines [
76]. COVID-19 vaccines have been the subject of several studies [
9,
10,
11,
12]. As a result of previous outbreaks of SARS CoV and MERS CoV, global industry leaders and researchers have attempted to develop a suitable vaccine [
77,
78]. The vaccine development approaches for SARS CoV2 include live attenuated virus [
79,
80,
81,
82], protein-based vaccines [
83,
84,
85,
86,
87,
88,
89,
90], viral vectors [
79,
91,
92,
93,
94], DNA vaccines [
95,
96,
97], mRNA vaccines [
98,
99,
100], and other self-assembling vaccines [
101]. The S protein/gene is the most preferred target site in SARS vaccine development, meaning the same strategy could potentially be useful in developing a SARS CoV2 vaccine [
102,
103].
2.1. Molecular Modeling of the Designed Multi-Epitopic Vaccines
Informatics tools are as convenient and cost-effective as in silico studies [
104,
105] and could help in designing a multi-epitope vaccine for COVID-19 treatments [
106,
107,
108,
109,
110,
111,
112,
113]. The potential peptide epitopes (antigenic and immunogenic) can help in facilitating the design of vaccines. The candidate vaccine constructs consist of distant epitopes. These epitopes are computed using B cells, cytotoxic T lymphocytes (CTL), and helper T lymphocytes (HTL) based on SARS CoV2 proteins [
114,
115]. The candidate vaccines are designed by fusing these epitopes with linkers [
116,
117,
118,
119]. These linkers help to stabilize the protein structure by producing a flexible conformation, protein folding, and functional domain separation. The binding affinity of the potential candidate vaccines with immune receptors was studied using the molecular docking approach.
2.1.1. Molecular Docking of the Construct SARS CoV2 Vaccine with the Related Antigenic Recognition Receptors (TLR-3, MHC-I, and MHC-II)
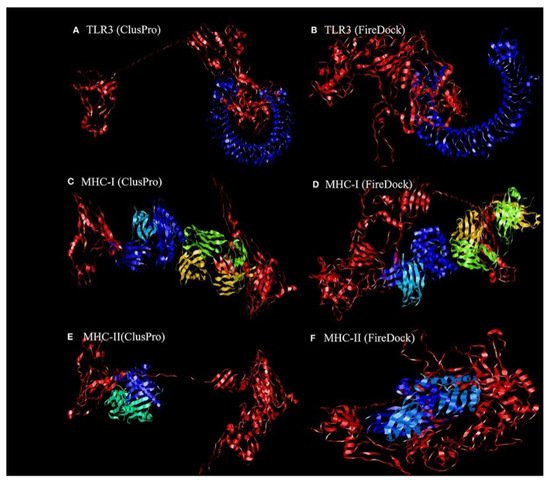
Dong et al. [
119] revealed the binding affinity between the candidate vaccines and antigenic recognition receptors of toll-like receptor-3 (TLR-3, 2A0Z) and the major histo compatibility complex (MHC-I, 4WUU), and MHC-II, 3C5J) present on the immune cell surfaces [
120], which were evaluated using molecular docking techniques. The ClusPro server,
https://cluspro.bu.edu/login.php?redir/queue.php (accessed on 20 October 2021), was used toperform the docking analysis. This server computed 44 models based on the electrostatic interaction and the desolvated energy. The server determined a total of twenty-nine model complexes each for TLR3, MHC-I, and MHC-II and the COVID-19 vaccine. The lowest binding energy scores of −1156.2, −1346.8, and −1309 kcal/mol were chosen and are represented in
Figure 2. The PatchDock server (
https://bioinfo3d.cs.tau.ac.il/PatchDock/ (accessed on 20 October 2021)) was used for further docking evaluation to confirm the binding affinity between the vaccine construct and the suggested receptors [
121]. The top 10 complexes were then refined using the FireDock algorithm [
121], which predicts the optimal complex with the help of energy functions. The best model complex of TLR3, MHC-I, and MHC-II receptors and the vaccine construct interaction (red color) showed global energy results, attractive van der Waals energy (VdW) results, repulsivevan der Waals (VdW) energy results, and atomic contact energy values (
Figure 2,
Table 1), verifying the stability of the docked complex.
Figure 2. The ligand–receptor docked complex:(
A,
C,
E) the docking interaction of the vaccine construct (red color) with the TLR-3, MHC-I, and MHC-II receptors (other colors), as assessed using ClusPro software;(
B,
D,
F)the docking interaction of the vaccine construct (red color) and TLR-3, MHC-I, and MHC-II receptors (other colors), as assessed using PatchDock [
119]. Permission and copyright © from Frontiers in Immunology (
www.frontiersin.org (accessed on 20 October 2021)).
Table 1. Static interactions of vaccines with TLR3, MHC-I, and MHC-II.
| Parameters |
TLR3 |
MHC-I |
MHC-II |
| lowest binding energy score (kcal/mol) |
−1156.2 |
−1346.8 |
−1309 |
| global energy |
−38.40 |
−22.97 |
−27.52 |
| attractive van der Waals energy (VdW) |
−26.02 |
−26.84 |
−26.86 |
| repulsive van der Waals energy (VdW) |
8.62 |
12.82 |
10.93 |
| atomic contact energy |
−11.06 |
−1.79 |
0.77 |
2.1.2. Molecular Docking of the SARS CoV2 Vaccine Construct with the Antigenic Recognition Receptors (ACE-2, TLR2, TLR4, HLA Alleles)
The candidate vaccines were designed and then tested with different epitopes [
122]. The construct vaccines, which have high antigenicity and are expected to produce high antibody titers, were combined with multi-epitope vaccines in order to enhance the immune response [
123,
124]. Three different construct vaccine constructs were used, with one comprising the top-scoring CD4 and CD8 epitopes lying in the S1 domain and the second one formed by taking two epitopes from the S1 domain and two from the S2 domain of the spike SARS CoV 2 protein, which represents the MHC-I and MHC-II binders. The last vaccine was created by combining a B-cell epitope with the second vaccine using a different adjuvant. The candidate vaccine constructs were also docked to different type of receptors, such as the ACE-2 receptor using PDB ID: 3sci, TLR2 with PDB ID: 2Z7X, TLR4 with PDB ID: 4G8A, B-cell receptor (BCR) CD79 using PDB ID: 3KG5 and HLA super family alleles; HLA A*02 01 using PDB ID: 4U6Y, the other HLA B*51 01 using PDB ID: 4MJI, and also class II HLA-DRB1*1402 using PDB ID: 6ATF [
122], representing broad-spectrum peptide-binding repertoires.
Molecular Docking of the Candidate Vaccines with ACE-2 Receptors
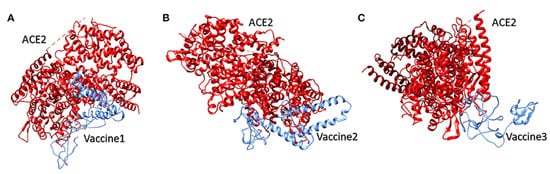
Vaccine 1 was docked with the ACE-2 receptor using the HADDOCK server (Guru Interface and refinement interface) [
125]. The server clustered 36 structures into seven different clusters. Vaccine 2 was also docked with the ACE-2 receptor, while the HADDOCK server clustered 18 structures into three clusters. While working on vaccine 3 and the ACE-2 receptor, the server clustered 22 structures into five clusters. The top cluster score, Z-score, and water-refined models are represented in
Figure 3. The latter (as shown in
Figure 3) represents docking interactions of the
h-ACE-2 protein complex (red) with the proposed multi-epitopic vaccines 1, 2, and 3 (blue), which were performed using the HADDOCKserver. The static interaction parameters of ACE-2 with the three construct vaccines 1,2, and 3 are displayed in
Table 2.
Figure 12. The docking interaction of the human ACE-2 protein complex (red) with the proposed multi-epitopic COVID-19 vaccines (
1–
3) representing in subfigures (
A–
C) (blue) respectively [
122]. Permission and copyright © from
www.frontiersin.org (accessed on 20 October 2021).
Table 2. Static interaction parameters of ACE-2 with vaccines 1, 2, and 3.
| Parameter |
ACE-2/Vaccine 1 |
ACE-2/Vaccine 2 |
ACE-2/Vaccine 3 |
| HADDOCK score (kcal/mol) |
39.8 +/− 29.1 |
0.3 +/− 9.8 |
147.5 +/− 15.0 |
| Z-score |
1.6 |
1.3 |
1.2 |
| water-refined models |
18% |
9.0% |
11% |
Molecular Docking of Vaccines with the Potential TLR2 and TLR4 Receptors
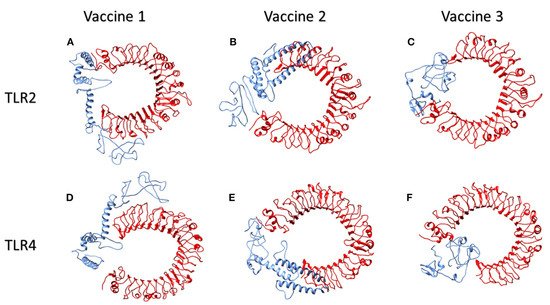
Toll-like receptors (TLR) TLR2 and TLR4, which are found on the cell surfaces, are activated by viral glycoproteins. They detect structural and non-structural proteins of the virus, suggesting the need to study cytokine release and inflammation. These TLR agonists have been shown to enhance the immune system’s response and viral clearance [
126]. TLR2 was docked with the three constructed vaccines. The interaction between vaccine 1 and TLR2 revealed 40 structures in six clusters. Similarly, the server clustered 80 structures into 12 clusters for the interaction of vaccine 2 and TLR2. Likewise, the HADDOCK server clustered 136 structures into 10 clusters; the top-scoring model, z- score, and the total water-refined model results from the interactions of TLR2 with vaccines 1, 2, and 3 are depicted in
Table 3. The three candidate vaccines also interacted with TLR4.In the case of the interaction of vaccine 1 and TLR4, the server clustered 157 structures into 13 clusters. Similarly, vaccine 2 interacted with the TLR4 receptor, clustering 47 structures into nine clusters. Likewise, the HADDOCK clustered 93 structures into eight clusters for the interaction between vaccine3 and TLR4. The top HADDOCK cluster scores with Z-values and the water-refined models are presented in
Table 3. The HADDOCK refinement interface was then used to refine the top cluster models. The 20 obtained structures were clustered into one single cluster. The final cluster consisted entirely of water-refined models. The observed statistics in the interaction of vaccines 1, 2, and 3 from their refined clusters were also studied [
122] and the complexes are shown in
Figure 13 and
Table 3.
Figure 13 illustrates the interactions of human TLR2 and TLR4 proteins (red) with the candidate multi-epitopic COVID-19 vaccines 1, 2, and 3 (blue).
Figure 4. The interactions of human TLR2 and TLR4 proteins (red) with the candidate multi-epitopic COVID-19 vaccines (
1-3) (blue) representing in subfigures (
A-F), respectively [
122]. Permission and copyright © from
www.frontiersin.org (accessed on 20 October 2021).
Table 3. Statics interaction parameters of TLR2 and TLR4 with vaccines 1, 2, and 3.
| Parameter |
Vaccine 1 |
Vaccine 2 |
Vaccine 3 |
| TLR2 |
TLR4 |
TLR2 |
TLR4 |
TLR2 |
TLR4 |
HADDOCK score
(kcal/mol) |
4. 2 +/− 20.8 |
37.9 +/− 7.8 |
23.7 +/− 12.1 |
16.8 +/− 23.4 |
16.7 +/− 14.0 |
23.3 +/− 5.7 |
| Z-score |
1.2 |
2.2 |
1.3 |
1.6 |
1.8 |
1.3 |
| water-refined models |
20.0% |
78.5 |
40.0% |
23.5 |
68.0% |
46.5 |
Molecular Modeling Interactions of the Suggested Vaccines with HLA Alleles
Studies showed that the docking analysis interaction of the proposed vaccine 1 and the HLA A allele [
122] with the water-refined models was59.0 percent. The HADDOCK server clustered 118 structures into 17 separate clusters. Likewise, the server clustered 97 structures into 17 clusters, depicting 48.5 percent of the water-refined models in the case of vaccine 2 and the HLA A allele. Similarly, the HADDOCK server clustered 187 structures into three clusters for vaccine 3, representing 93.5 percent of the water-refined models. The best HADDOCK results with z-scores are represented in
Table 4. Here, 115 structures were clustered into 15 separate clusters using HADDOCK server for the interaction between vaccine 1 and the HLA B allele, representing a total of 57.5 percent of the water-refined models. Additionally, HADDOCK clustered 84 structures into nine clusters for the interaction of vaccine2 and the HLA B allele, depicting 42 percent of the water-refined models. The HADDOCK server aggregated 168 likely structures into 10 clusters representing 84 percent of the water-refined models for vaccine 3. The top-scoring cluster and the static parameters are depicted in
Table 4. Moreover, when vaccine 1 was docked into the HLA DRB1 allele, the HADDOCK server showed 67 probable structures clustered into different clusters, accounting for 33.5 percent of the water-refined models. Likewise, the HADDOCK server clustered 64 structures into 11 different clusters for vaccine 2 and the HLA DRB1 allele, representing32 percent of the water-refined models. HADDOCK clustered 93 probable structures into 13 different clusters for vaccine 3, representing 46.5 percent of the water-refined models. The scoring parameters are shown in
Table 4. The HADDOCK refinement interface refined and then clustered the top cluster model from 20 structures into one single cluster, representing 100 percent of the water-refined models. The observed statistics for the interactions of vaccines 1, 2, and 3 with their respective refined clusters were also studied [
122]. Finally, the vaccine mRNA was improved using the Java Codon Adaptation Tool in order to ensure that the designed vaccines were effective in a specific expression system.
Table 4. Static interaction parameters of HLA A alleles, HLA Balleles, and HLA DRB1 allele with vaccines 1, 2, and 3.
| Parameter |
Vaccine 1 |
Vaccine 2 |
Vaccine 3 |
| HLA A Allele |
HLA B Allele |
HLA DRB1 Allele |
HLA A Alleles |
HLA B Alleles |
HLA DRB1 allele |
HLA A Alleles |
HLA B Alleles |
HLA DRB1 Allele |
HADDOCK score
(kcal/mol) |
26.5 +/− 2.7 |
57.5 +/− 12.8 |
27.8 +/− 6.0 |
57.5 +/− 12.8 |
18.7 +/− 8.7 |
24.8 +/− 25.6 |
34.7 +/− 1.9 |
41.2 +/− 18.7 |
37.1 +/− 11. 8 |
| Z-score |
2.5 |
2.3 |
2.3 |
2.3 |
1.6 |
1.7 |
1.1 |
2.1 |
−1.5 |
| water-refined models |
59.0% |
57.5 |
33.5 |
48.5 |
42 |
32 |
93.5 |
84 |
46.5 |
This entry is adapted from the peer-reviewed paper 10.3390/coatings11111273