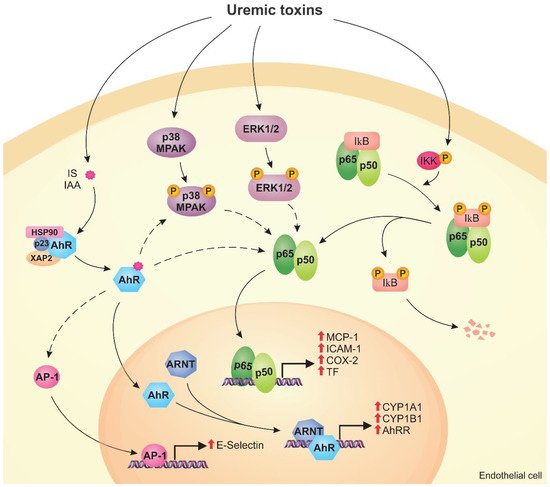
Uremic toxins can induce endothelial dysfunction in patients with chronic kidney disease (CKD). Indeed, the structure of the endothelial monolayer is damaged in CKD, and studies have shown that the uremic toxins contribute to the loss of cell–cell junctions, increasing permeability. Membrane proteins, such as transporters and receptors, can mediate the interaction between uremic toxins and endothelial cells. In these cells, uremic toxins induce oxidative stress and activation of signaling pathways, including the aryl hydrocarbon receptor (AhR), nuclear factor kappa B (NF-κB), and mitogen-activated protein kinase (MAPK) pathways. The activation of these pathways leads to overexpression of proinflammatory (e.g., monocyte chemoattractant protein-1, E-selectin) and prothrombotic (e.g., tissue factor) proteins. Uremic toxins also induce the formation of endothelial microparticles (EMPs), which can lead to the activation and dysfunction of other cells, and modulate the expression of microRNAs that have an important role in the regulation of cellular processes.
- uremic toxins
- endothelium
- endothelial dysfunction
1. Introduction
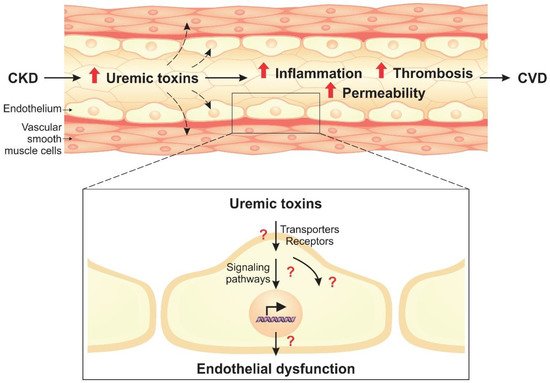
2. Cell Signaling Pathways Altered by Uremic Toxins
2.1. AhR Pathway
2.2. NF-κB Pathway
2.3. MAPK Pathway
3. Modulation of MicroRNAs by Uremic Toxins in Endothelial Cells
4. Uremic Toxins Induce the Formation of Endothelial Microparticles
This entry is adapted from the peer-reviewed paper 10.3390/toxins12060412
References
- Jing, Y.J.; Ni, J.W.; Ding, F.H.; Fang, Y.H.; Wang, X.Q.; Wang, H.B.; Chen, X.N.; Chen, N.; Zhan, W.W.; Lu, L.; et al. p-Cresyl sulfate is associated with carotid arteriosclerosis in hemodialysis patients and promotes atherogenesis in apoE−/− mice. Kidney Int. 2016, 89, 439–449.
- Wang, C.H.; Lai, Y.H.; Kuo, C.H.; Lin, Y.L.; Tsai, J.P.; Hsu, B.G. Association between serum indoxyl sulfate levels and endothelial function in non-dialysis chronic kidney disease. Toxins 2019, 11, 589.
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Serum Indoxyl Sulfate Is Associated with Vascular Disease and Mortality in Chronic Kidney Disease Patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558.
- Wu, I.W.; Hsu, K.H.; Hsu, H.J.; Lee, C.C.; Sun, C.Y.; Tsai, C.J.; Wu, M.S. Serum free p-cresyl sulfate levels predict cardiovascular and all-cause mortality in elderly hemodialysis patients—A prospective cohort study. Nephrol. Dial. Transplant. 2012, 27, 1169–1175.
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z. A free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol. Dial. Transplant. 2010, 25, 1183–1191.
- Rattazzi, M.; Villalta, S.; De Lucchi, L.; Sponchiado, A.; Galliazzo, S.; Faggin, E.; Pagliara, V.; Zilli, C.; Callegari, E.; Caberlotto, L.; et al. Chronic kidney disease is associated with increased risk of venous thromboembolism recurrence. Thromb. Res. 2017, 160, 32–37.
- Betriu, A.; Martinez-Alonso, M.; Arcidiacono, M.V.; Cannata-Andia, J.; Pascual, J.; Valdivielso, J.M.; Fernandez, E. Prevalence of subclinical atheromatosis and associated risk factors in chronic kidney disease: The NEFRONA study. Nephrol. Dial. Transplant. 2014, 29, 1415–1422.
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G. Biochemical and Clinical Impact of Organic Uremic Retention Solutes: A Comprehensive Update. Toxins 2018, 10, 33.
- Boelaert, J.; Lynen, F.; Glorieux, G.; Schepers, E.; Neirynck, N.; Vanholder, R. Metabolic profiling of human plasma and urine in chronic kidney disease by hydrophilic interaction liquid chromatography coupled with time-of-flight mass spectrometry: A pilot study. Anal. Bioanal. Chem. 2017, 409, 2201–2211.
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270.
- Okuno, S.; Ishimura, E.; Kohno, K.; Fujino-Katoh, Y.; Maeno, Y.; Yamakawa, T.; Inaba, M.; Nishizawa, Y. Serum β2-microglobulin level is a significant predictor of mortality in maintenance haemodialysis patients. Nephrol. Dial. Transplant. 2009, 24, 571–577.
- De Oliveira, R.B.; Liabeuf, S.; Okazaki, H.; Lenglet, A.; Desjardins, L.; Lemke, H.-D.; Vanholder, R.; Choukroun, G.; Massy, Z.A. The clinical impact of plasma leptin levels in a cohort of chronic kidney disease patients. Clin. Kidney J. 2013, 6, 63–70.
- Maciel, R.; Cunha, R.; Busato, V.; Franco, C.; Gregório, P.; Dolenga, C.; Nakao, L.; Massy, Z.; Boullier, A.; Pecoits-Filho, R.; et al. Uremia Impacts VE-Cadherin and ZO-1 Expression in Human Endothelial Cell-to-Cell Junctions. Toxins 2018, 10, 404.
- Favretto, G.; Souza, L.M.; Gregório, P.C.; Cunha, R.S.; Maciel, R.A.P.; Sassaki, G.L.; Toledo, M.G.; Pecoits-Filho, R.; Souza, W.M.; Stinghen, A.E.M. Role of Organic Anion Transporters in the Uptake of Protein-Bound Uremic Toxins by Human Endothelial Cells and Monocyte Chemoattractant Protein-1 Expression. J. Vasc. Res. 2017, 54, 170–179.
- Soshilov, A.A.; Motta, S.; Bonati, L.; Denison, M.S. Transitional States in Ligand-Dependent Transformation of the Aryl Hydrocarbon Receptor into Its DNA-Binding Form. Int. J. Mol. Sci. 2020, 21, 2474.
- Addi, T.; Poitevin, S.; McKay, N.; El Mecherfi, K.E.; Kheroua, O.; Jourde-Chiche, N.; de Macedo, A.; Gondouin, B.; Cerini, C.; Brunet, P.; et al. Mechanisms of tissue factor induction by the uremic toxin indole-3 acetic acid through aryl hydrocarbon receptor/nuclear factor-kappa B signaling pathway in human endothelial cells. Arch. Toxicol. 2019, 93, 121–136.
- Watanabe, I.; Tatebe, J.; Namba, S.; Koizumi, M.; Yamazaki, J.; Morita, T. Activation of Aryl Hydrocarbon Receptor Mediates Indoxyl Sulfate-Induced Monocyte Chemoattractant Protein-1 Expression in Human Umbilical Vein Endothelial Cells. Circ. J. 2013, 77, 224–230.
- Ambolet-Camoit, A.; Ottolenghi, C.; Leblanc, A.; Kim, M.J.; Letourneur, F.; Jacques, S.; Cagnard, N.; Guguen-Guillouzo, C.; Barouki, R.; Aggerbeck, M. Two persistent organic pollutants which act through different xenosensors (alpha-endosulfan and 2,3,7,8 tetrachlorodibenzo-p-dioxin) interact in a mixture and downregulate multiple genes involved in human hepatocyte lipid and glucose metabolism. Biochimie 2015, 116, 79–91.
- Han, S.G.; Han, S.S.; Toborek, M.; Hennig, B. EGCG protects endothelial cells against PCB 126-induced inflammation through inhibition of AhR and induction of Nrf2-regulated genes. Toxicol. Appl. Pharmacol. 2012, 261, 181–188.
- Asai, H.; Hirata, J.; Watanabe-Akanuma, M. Indoxyl glucuronide, a protein-bound uremic toxin, inhibits hypoxia-inducible factor‒dependent erythropoietin expression through activation of aryl hydrocarbon receptor. Biochem. Biophys. Res. Commun. 2018, 504, 538–544.
- Kolachalama, V.B.; Shashar, M.; Alousi, F.; Shivanna, S.; Rijal, K.; Belghasem, M.E.; Walker, J.; Matsuura, S.; Chang, G.H.; Gibson, C.M.; et al. Uremic Solute-Aryl Hydrocarbon Receptor-Tissue Factor Axis Associates with Thrombosis after Vascular Injury in Humans. J. Am. Soc. Nephrol. 2018, 29, 1063–1072.
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9.
- Ito, S.; Osaka, M.; Edamatsu, T.; Itoh, Y.; Yoshida, M. Crucial Role of the Aryl Hydrocarbon Receptor (AhR) in Indoxyl Sulfate-Induced Vascular Inflammation. J. Atheroscler. Thromb. 2016, 23, 960–975.
- Dou, L.; Sallée, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. Soc. Nephrol. 2015, 26, 876–887.
- Gondouin, B.; Cerini, C.; Dou, L.; Sallée, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744.
- Assefa, E.G.; Yan, Q.; Gezahegn, S.B.; Salissou, M.T.M.; He, S.; Wu, N.; Zuo, X.; Ying, C. Role of resveratrol on indoxyl sulfate-induced endothelial hyperpermeability via aryl hydrocarbon receptor (AHR)/Src-dependent pathway. Oxid. Med. Cell. Longev. 2019, 2019.
- Koizumi, M.; Tatebe, J.; Watanabe, I.; Yamazaki, J.; Ikeda, T.; Morita, T. Aryl Hydrocarbon Receptor Mediates Indoxyl Sulfate-Induced Cellular Senescence in Human Umbilical Vein Endothelial Cells. J. Atheroscler. Thromb. 2014, 21, 904–916.
- Jansen, J.; Jansen, K.; Neven, E.; Poesen, R.; Othman, A.; van Mil, A.; Sluijter, J.; Sastre Torano, J.; Zaal, E.A.; Berkers, C.R.; et al. Remote sensing and signaling in kidney proximal tubules stimulates gut microbiome-derived organic anion secretion. Proc. Natl. Acad. Sci. USA 2019, 116, 16105–16110.
- Machado, T.S.; Poitevin, S.; Paul, P.; McKay, N.; Jourde-Chiche, N.; Legris, T.; Mouly-Bandini, A.; Dignat-George, F.; Brunet, P.; Masereeuw, R.; et al. Indoxyl sulfate upregulates liver P-glycoprotein expression and activity through aryl hydrocarbon receptor signaling. J. Am. Soc. Nephrol. 2018, 29, 906–918.
- Dou, L.; Poitevin, S.; Sallée, M.; Addi, T.; Gondouin, B.; McKay, N.; Denison, M.S.; Jourde-Chiche, N.; Duval-Sabatier, A.; Cerini, C.; et al. Aryl hydrocarbon receptor is activated in patients and mice with chronic kidney disease. Kidney Int. 2018, 93, 986–999.
- Shivanna, S.; Kolandaivelu, K.; Shashar, M.; Belghasim, M.; Al-Rabadi, L.; Balcells, M.; Zhang, A.; Weinberg, J.; Francis, J.; Pollastri, M.P.; et al. The Aryl Hydrocarbon Receptor is a Critical Regulator of Tissue Factor Stability and an Antithrombotic Target in Uremia. J. Am. Soc. Nephrol. 2016, 27, 189–201.
- Lano, G.; Laforêt, M.; Von Kotze, C.; Perrin, J.; Addi, T.; Brunet, P.; Poitevin, S.; Burtey, S.; Dou, L. Aryl Hydrocarbon Receptor Activation and Tissue Factor Induction by Fluid Shear Stress and Indoxyl Sulfate in Endothelial Cells. Int. J. Mol. Sci. 2020, 21, 2392.
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85.
- Sun, S.-C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558.
- Masai, N.; Tatebe, J.; Yoshino, G.; Morita, T. Indoxyl sulfate stimulates monocyte chemoattractant protein-1 expression in human umbilical vein endothelial cells by inducing oxidative stress through activation of the NADPH oxidase-nuclear factor-kappaB pathway. Circ. J. 2010, 74, 2216–2224.
- Tumur, Z.; Shimizu, H.; Enomoto, A.; Miyazaki, H.; Niwa, T. Indoxyl Sulfate Upregulates Expression of ICAM-1 and MCP-1 by Oxidative Stress-Induced NF-ĸB Activation. Am. J. Nephrol. 2010, 31, 435–441.
- Buendía, P.; Carracedo, J.; Soriano, S.; Madueño, J.A.; Ortiz, A.; Martín-Malo, A.; Aljama, P.; Ramírez, R. Klotho Prevents NFκB Translocation and Protects Endothelial Cell From Senescence Induced by Uremia. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 1198–1209.
- Abbasian, N.; Burton, J.O.; Herbert, K.E.; Tregunna, B.-E.; Brown, J.R.; Ghaderi-Najafabadi, M.; Brunskill, N.J.; Goodall, A.H.; Bevington, A. Hyperphosphatemia, Phosphoprotein Phosphatases, and Microparticle Release in Vascular Endothelial Cells. J. Am. Soc. Nephrol. 2015, 26, 2152–2162.
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. p38MAPK: Stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009, 15, 369–379.
- Morrison, D.K. MAP Kinase Pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254.
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83.
- Shang, F.; Wang, S.-C.; Hsu, C.-Y.; Miao, Y.; Martin, M.; Yin, Y.; Wu, C.-C.; Wang, Y.-T.; Wu, G.; Chien, S.; et al. MicroRNA-92a Mediates Endothelial Dysfunction in CKD. J. Am. Soc. Nephrol. 2017, 28, 3251–3261.
- Kumar, S.; Kim, C.W.; Simmons, R.D.; Jo, H. Role of Flow-Sensitive microRNAs in Endothelial Dysfunction and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2206–2216.
- Schober, A.; Weber, C. Mechanisms of MicroRNAs in Atherosclerosis. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 583–616.
- Wiese, C.B.; Zhong, J.; Xu, Z.-Q.; Zhang, Y.; Ramirez Solano, M.A.; Zhu, W.; Linton, M.F.; Sheng, Q.; Kon, V.; Vickers, K.C. Dual inhibition of endothelial miR-92a-3p and miR-489-3p reduces renal injury-associated atherosclerosis. Atherosclerosis 2019, 282, 121–131.
- Kétszeri, M.; Kirsch, A.; Frauscher, B.; Moschovaki-Filippidou, F.; Mooslechner, A.A.; Kirsch, A.H.; Schabhuettl, C.; Aringer, I.; Artinger, K.; Pregartner, G.; et al. MicroRNA-142-3p improves vascular relaxation in uremia. Atherosclerosis 2019, 280, 28–36.
- Li, S.; Xie, Y.; Yang, B.; Huang, S.; Zhang, Y.; Jia, Z.; Ding, G.; Zhang, A. MicroRNA-214 targets COX-2 to antagonize indoxyl sulfate (IS)-induced endothelial cell apoptosis. Apoptosis 2020, 25, 92–104.
- Fourdinier, O.; Schepers, E.; Metzinger-Le Meuth, V.; Glorieux, G.; Liabeuf, S.; Verbeke, F.; Vanholder, R.; Brigant, B.; Pletinck, A.; Diouf, M.; et al. Serum levels of miR-126 and miR-223 and outcomes in chronic kidney disease patients. Sci. Rep. 2019, 9, 4477.
- M’baya-Moutoula, E.; Marchand, A.; Six, I.; Bahrar, N.; Celic, T.; Mougenot, N.; Maitrias, P.; Massy, Z.A.; Lompreh, A.-M.; Metzinger, L.; et al. Inhibition of miR-223 expression using a sponge strategy decreases restenosis in rat injured carotids. Curr. Vasc. Pharmacol. 2019, 17.
- Metzinger-Le Meuth, V.; Metzinger, L. miR-223 and other miRNA’s evaluation in chronic kidney disease: Innovative biomarkers and therapeutic tools. Non-Coding RNA Res. 2019, 4, 30–35.
- Favretto, G.; Cunha, R.S.d.; Dalboni, M.A.; Oliveira, R.B.d.; Barreto, F.d.C.; Massy, Z.A.; Stinghen, A.E.M. Endothelial Microparticles in Uremia: Biomarkers and Potential Therapeutic Targets. Toxins 2019, 11, 267.
- Carmona, A.; Guerrero, F.; Buendia, P.; Obrero, T.; Aljama, P.; Carracedo, J. Microvesicles Derived from Indoxyl Sulfate Treated Endothelial Cells Induce Endothelial Progenitor Cells Dysfunction. Front. Physiol. 2017, 8, 666.
- Ryu, J.H.; Jeon, E.Y.; Kim, S.J. Indoxyl sulfate-induced extracellular vesicles released from endothelial cells stimulate vascular smooth muscle cell proliferation by inducing transforming growth factor-beta production. J. Vasc. Res. 2019, 56, 129–138.
- Soriano, S.; Carmona, A.; Triviño, F.; Rodriguez, M.; Alvarez-Benito, M.; Martín-Malo, A.; Alvarez-Lara, M.-A.; Ramírez, R.; Aljama, P.; Carracedo, J. Endothelial damage and vascular calcification in patients with chronic kidney disease. Am. J. Physiol. Physiol. 2014, 307, F1302–F1311.
- Gao, C.; Xie, R.; Yu, C.; Ma, R.; Dong, W.; Meng, H.; Zhang, Y.; Si, Y.; Zhang, Z.; Novakovic, V.; et al. Thrombotic role of blood and endothelial cells in uremia through phosphatidylserine exposure and microparticle release. PLoS ONE 2015, 10, 1–16.
- Meijers, B.K.I.; Van kerckhoven, S.; Verbeke, K.; Dehaen, W.; Vanrenterghem, Y.; Hoylaerts, M.F.; Evenepoel, P. The Uremic Retention Solute p-Cresyl Sulfate and Markers of Endothelial Damage. Am. J. Kidney Dis. 2009, 54, 891–901.
- Faure, V.; Dou, L.; Sabatier, F.; Cerini, C.; Sampol, J.; Berland, Y.; Brunet, P.; Dignat-George, F. Elevation of circulating endothelial microparticles in patients with chronic renal failure. J. Thromb. Haemost. 2006, 4, 566–573.
- Di Marco, G.S.; König, M.; Stock, C.; Wiesinger, A.; Hillebrand, U.; Reiermann, S.; Reuter, S.; Amler, S.; Köhler, G.; Buck, F.; et al. High phosphate directly affects endothelial function by downregulating annexin II. Kidney Int. 2013, 83, 213–222.
- Amabile, N.; Guerin, A.P.; Leroyer, A.P.; Mallat, Z.; Nguyen, C.; Boddaert, J.; London, G.M.; Tedgui, A.; Boulanger, C.M. Circulating Endothelial Microparticles Are Associated with Vascular Dysfunction in Patients with End-Stage Renal Failure. J. Am. Soc. Nephrol. 2005, 16, 3381–3388.
- Mörtberg, J.; Lundwall, K.; Mobarrez, F.; Wallén, H.; Jacobson, S.H.; Spaak, J. Increased concentrations of platelet- and endothelial-derived microparticles in patients with myocardial infarction and reduced renal function- a descriptive study. BMC Nephrol. 2019, 20, 71.
- Jalal, D.; Renner, B.; Laskowski, J.; Stites, E.; Cooper, J.; Valente, K.; You, Z.; Perrenoud, L.; Le Quintrec, M.; Muhamed, I.; et al. Endothelial Microparticles and Systemic Complement Activation in Patients With Chronic Kidney Disease. J. Am. Heart Assoc. 2018, 7, e007818.
- Ryu, J.-H.; Park, H.; Kim, S.-J. The effects of indoxyl sulfate-induced endothelial microparticles on neointimal hyperplasia formation in an ex vivo model. Ann. Surg. Treat. Res. 2017, 93, 11.