Triple-negative breast cancer is a combative cancer type with a highly inflated histological grade that leads to poor theragnostic value. Gene, protein, and receptor-specific targets have shown effective clinical outcomes in patients with TNBC. Cells are frequently exposed to DNA-damaging agents. DNA damage is repaired by multiple pathways; accumulations of mutations occur due to damage to one or more pathways and lead to alterations in normal cellular mechanisms, which lead to development of tumors. Advances in target-specific cancer therapies have shown significant momentum; most treatment options cause off-target toxicity and side effects on healthy tissues. PARP (poly(ADP-ribose) polymerase) is a major protein and is involved in DNA repair pathways, base excision repair (BER) mechanisms, homologous recombination (HR), and nonhomologous end-joining (NEJ) deficiency-based repair mechanisms. DNA damage repair deficits cause an increased risk of tumor formation. Inhibitors of PARP favorably kill cancer cells in BRCA-mutations. For a few years, PARPi has shown promising activity as a chemotherapeutic agent in BRCA1- or BRCA2-associated breast cancers, and in combination with chemotherapy in triple-negative breast cancer.
1. Introduction
Breast cancer (BC) is the most common cancer that occurs in women worldwide [
1]. BC is caused by accumulation of somatic mutations in breast cells, which impair cell division and DNA repair mechanisms, resulting in irregular cell growth proliferation, differentiation, and ultimately, progression of tumorigenesis [
2,
3]. Triple-negative breast cancer is more belligerent and has a poorer prognosis than other types of breast cancer. Triple-negative breast cancer (TNBC) accounts for approximately 15% of all BC, and lacks human epidermal growth factor receptor 2 (HER2), progesterone receptor (PR), and estrogen receptor (ER) expression and amplification. If we compare it with another type of BC, TNBC exhibits inherently aggressive clinical symptoms and poorer clinical outcomes [
4,
5,
6,
7]. Presently, the clinical targeted drugs for BC include poly-(ADP)-ribose polymerase (PARP) inhibitors (PARPi), CDK4/6 inhibitors (CDK4/6i), PI3K inhibitors, and AKT inhibitors—but none of these drugs alone are very effective against TNBC [
8]. There is an urgent need for the rational exploration of drug compatibility and potential targets for TNBC [
7,
8]. PARP1 (poly (ADP-ribose) polymerase 1) was discovered approximately 50 years ago and is involved in gene transcription, DNA repair, and cell death [
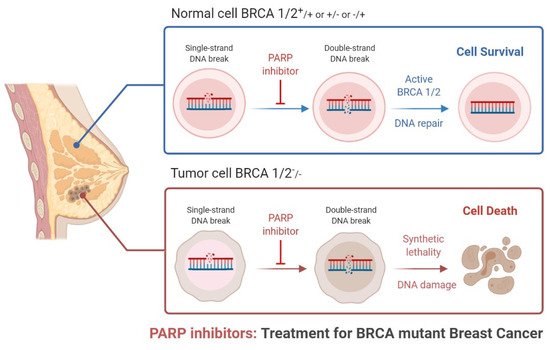
9]. PARP1 has acceptable therapeutic importance against cancer, as shown in
Figure 1 and
Figure 2 [
10]. PARP inhibitors have emerged as effective treatments in clinical trials for sporadic TNBC and
BRCA-associated cancers [
11]. There are various types of PARP inhibitors under clinical trial such as olaparib, BSI-201, talazoparib, rucaparib veliparib, and niraparib [
10,
11]. Inhibition of the PARP-1 and PARP-2 enzymes is believed to be attained mainly via binding of the NAD+ catalytic domain side chain, extending out of the NAD catalytic site of the PARP inhibitor [
12]. It also thought that the PARP enzyme locks on to the site of DNA damage, preventing its usual release from DNA molecules [
10,
11,
12,
13,
14,
15]. PARP-1 binds to the damaged site through its zinc-finger domains in the presence of SS (single-stranded)-DNA breaks [
13]. PARP-1 and poly (ADP) polymerization recruits and binds other DNA-repair proteins, leading under normal cell physiology to an increasingly negative charge on the enzyme, and eventual dissociation from the DNA [
14]. Some clinical investigations have shown the need for HRD (homologous recombination DNA repair) in facilitating PARP inhibition, via loss of
BRCA function [
16,
17]. Researchers from the field have suggested that PARP inhibition is associated with the induction of DNA damage by chemotherapy in the more general cohort of TNBC.
Figure 1. Role of PARP inhibitors in treatments for BRCA mutant breast cancer.
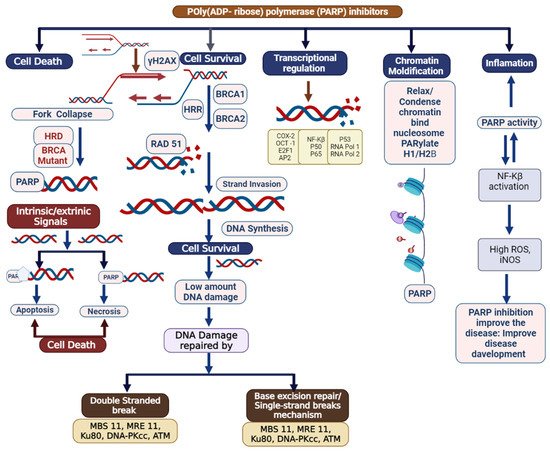
Figure 2. Schematic delineating the multifaceted nature of poly (ADP) ribose polymerase (PARP): DNA repair, chromatin modification, inflammation, transcriptional regulation, and cell death. Potential role of elevated PARP-1 in tumorigenesis. After DNA damage, PARP-1 activates DNA repair. However, PARP-1 also acts as a co-activator of NFkB signaling, which can propagate inflammatory signaling and lead to more DNA damage, including the formation of oxidatively clustered DNA lesions (OCDLs). The formation of OCDLs is elevated in numerous tumor types. PARP-1 activity could potentially be beneficial or harmful in the repair of ROS-induced DNA lesions.
2. Clinical Applications of PARP Inhibitors in TNBC
PARP inhibitors have been shown to have effective clinical outcomes against different types of cancer. There are various clinical trials registered investigating PARPi therapies (Table 1, Table 2 and Table 3).
2.1. Olaparib
Olaparib is a potent oral PARP inhibitor effective against
BRCA1 and
BRCA2 mutations [
29]. A multicentric clinical evaluation of olaparib was carried out using it as a monotherapy for inpatients with germline
BRCA1/2 mutations [
56,
62,
63]. Olaparib was administered to the patient twice a day at a dose of 400 mg. The clinical trial was performed with 298 patients, out of which, effective clinical therapy was observed in 12.9%, with adverse effects of vomiting, nausea, and fatigue observed [
64,
65,
66,
67,
68]. Another study was performed to optimize the drug concentration and determine its maximum dose and minimum dose. In patients with
BRCA1 and
BRCA2 mutations, an ORR (overall response rate) of 11 (41%) was observed in 27 patients with a 400 mg dose twice daily. An ORR of 6 (22%) was observed in 27 patients with 100 mg doses twice daily; the ORR was observed to be 7/13 (54%) with higher doses and 4/16 (25%) with lower doses in TNBC patients. In higher dose-tested patients, some adverse effects were observed, such as anemia, vomiting, nausea, and fatigue. Olaparib was approved by the FDA based on the clinical outcomes of the patient [
65,
66,
67,
68]. A phase 3 clinical trial employed olaparib monotherapy in germline
BRCA mutations with
HER2 negativity and, at minimum, previous chemotherapy therapy [
67]. A total of 300 patients were selected randomly in a 2:1 ratio into two groups; group one was administered 300 mg olaparib twice daily, and 92 patients in group two were administered vinorelbine or capecitabine and eribulin in 21-day cycles. Out of 300, 49.8% of the TNBC patients were included in the olaparib group and 49.5% of the TNBC patients received standard therapy [
3,
64,
65,
66]. Median PFS was significantly longer in the olaparib group than in the standard therapy group (7.0 months vs. 4.2 months; hazard ratio for disease progression or death, 0.58; 95% CI, 0.43 to 0.80;
p < 0.001). In the subgroup analysis, the hazard ratio for PFS was 0.43 (95% CI, 0.29–0.63) for patients with TNBC [
3,
66,
67,
68]. The response rate was 59.9% in the olaparib group and 28.8% in the standard therapy group, while the rate of grade 3 or higher adverse events was 36.6% in the olaparib group and 50.5% in the standard therapy group; the rate of treatment discontinuation due to toxic effects was 4.9% and 7.7%, respectively [
66,
67,
68,
69]. Metabolism of olaparib occurs via oxidation and dehydrogenation and does so progressively via the use of other factors such as sulfate conjugate and glucuronide [
65,
66]. Olaparib is mainly excreted through urine (44%) and feces (42%) [
66].
OlympiAD was a randomized open clinical phase III trial (NCT02000622) assessing the daily administration of 600 mg olaparib tablets. A total of 302 patients who had received two or fewer prior treatments were randomized in a 2:1 ratio to olaparib or chemotherapy. The results showed significantly prolonged PFS with olaparib versus standard therapy (7.0 vs. 4.2 months; hazard ratio (HR), 0.58; 95% CI, 0.43–0.8;
p < 0.001); Response rates were observed to be 59.9% vs. 28.8% (olaparib vs. standard group) [
64]. Olaparib was the first PARP inhibitor to establish higher efficacy and tolerability than standard chemotherapy in g
BRCA-mutated advanced BC [
65,
66,
67,
68]. According to earlier results, the FDA approved olaparib as the first PARP inhibitor for the treatment of this patient subgroup. However, in the interim analysis, no differences in overall survival (OS) were observed between the two groups [
68,
69,
70]. The 3-year OS was 40.8% versus 12.8% in the two groups, respectively, in patients with TNBC. Currently, research on PARP inhibitors for adjuvant therapy and neoadjuvant therapy, as well as for the prevention of BC, is ongoing—including the OlympiA (phase III) and GeparSixto studies; in the future, the results of these studies will evaluate adjuvant therapy with olaparib for
HER-2-/g
BRCAm BC and explore the value of a PARP inhibitor in neoadjuvant therapy, respectively [
71,
72,
73]. Various remarkable drugs have been approved to benefit patients with TNBC, including the PARP inhibitors olaparib and talazoparib for germline
BRCA mutation-associated breast cancer (g
BRCAm-BC) and immunotherapy using the checkpoint inhibitor atezolizumab, in combination with nab-paclitaxel for programmed cell death-ligand 1-positive (PD-L1+) advanced TNBC [
66,
73].
2.2. Iniparib (BSI-201)
Iniparib was the first potent PARP1 inhibitor, effective against cancer cell lines with 40–128 μM IC50 values, and is not toxic at 200 mg/kg in Syrian hamsters [
74,
75]. The efficacy of iniparib (BSI-201) was established by caspase-3 and TUNEL staining of OVCAR-3 tumors; iniparib efficacy was high in combination with topotecan [
70,
71,
72]. Iniparib used together with a PARP-1 inhibitor has also shown efficacy in DNA repair mechanisms [
72]. One clinical trial investigated patients with metastatic TNBC [
74,
75], in which a total of 123 patients were selected randomly and two groups were made; in each group, patients received 1000 mg/m
2 gemcitabine and carboplatin on days 1 and 8, either with or without 5.6 mg/kg iniparib on days 1, 4, 8, and 11, over a cycle of 21 days [
74,
76]. The clinical efficacy of iniparib was increased with carboplatin and gemcitabine, and the ORR was increased from 32% to 52%. The time duration of the iniparib dose was also increased from a median PFS of 3.6 months to 5.9 months, and the median ORR from 7.7 months to 12.3 months; the hazard ratio for death was observed to be 0.57;
p = 0.01. ORR and PFS were analyzed further in a phase III clinical trial; the trial did not find successful treatment of patients [
76].
2.3. Niraparib
Niraparib is a PARP1 and PARP1 inhibitor. Niraparib is indicated as a maintenance treatment for recurrent cancer patients, mainly with HR deficiency (HRD) with positive status [
73,
77]. HRD has been linked to deleterious
BRCA mutations in patients, with disease development occurring more than six months later following platinum-based chemotherapy [
73,
77]. Niraparib was extended for use in the care treatment of adults following first-line platinum-based chemotherapy [
67,
73].
Patients with solid tumors (
BRCA1 or
BRCA2 mutation carrier) were enrolled in a phase I clinical trial [
76,
77,
78,
79]. The currently used therapeutic option was tested along with niraparib in
BRCA-mutated metastatic breast cancer; patients with germline
BRCA mutations were treated with a PARP inhibitor rather than chemotherapy, and the availability of PARP inhibitors increased [
76,
77,
78,
79]. No safety concerns have been noted by the IDMC (Independent Data Monitoring Committee) concerning niraparib [
76,
77,
78,
79]. The clinical outcome from the BRAVO (Breast Cancer Risk and Various Outcomes) trial is expected to be supportive of a planned trial of niraparib in combination with an anti-PD-1 antibody in women with metastatic TNBC [
76,
77,
78,
79].
2.4. Veliparib (ABT-888)
Veliparib (ABT-888) is a potent PARP1 and PARP2 inhibitor used as a neoadjuvant. It has good pharmacokinetic properties and has shown effective clinical outcomes [
80]. Veliparib is effective in platinum-based therapy in xenograft models [
80,
81]. Significantly, the eradication of solid tumors following neoadjuvant chemotherapy, designated the clinical–pathological response in breast and axillary nodes during surgery, is connected with progression-free survival (PFS) and overall survival rates (OSRs)—with strong correlations in TNBC and
HER2-positive disease, raising interest in the neoadjuvant approach [
82,
83]. Veliparib was clinically evaluated in TNBC patients in combination with carboplatin; it was also tested against the NAD+ catalytic enzyme SIRT2, showing inactivity against >5000 nM of the enzyme. Receptor-binding assays were performed in 74 patients for Veliparib receptor profile analysis at a concentration of 10 μM [
81,
82,
83]. Multiple investigations were carried out, such as control-specific binding at 50% of human 5-HT7 (84%) sites with an IC50s value of 1.2 μM; IC50s at H1(61%), with an IC value of 5.3 μM; and human 5-HT1A (91%) with a IC50s value of 1.5 μM. c-Met knockdown cells show shMet-A (95% CI = 4–4.5) tumor growth retardation with up to 60 μM Veliparib (ABT-888) [
81,
82,
83]. When treated with 38 μM Veliparib, c-Met knockdown cells show shMet-B (95% CI = 1.3–2.5) tumor growth inhibition. Cell viability was higher with 1,000 µM sulfur mustard (SM) exposure in HaCaT cells at 6 h post-treatment by Veliparib [
81,
82,
83]. Additionally, Veliparib no longer shows protective effects at 24 h post SM exposure.
Randomized patients were selected to receive either paclitaxel as monotherapy or veliparib and carboplatin as a combination therapy, followed by doxorubicin and cyclophosphamide given in four cycles [
83]. Clinical outcomes were examined, with estimated rates of PCR of 51% in the combination group with TNBC patients and 26% in the control group of patients [
83]. For the phase III clinical trial, 634 patients were selected based on histological clinical stage II–III TNBC with no previous therapy for potentially curative surgery—they were randomly assigned to two groups; group I was treated with 50 mg veliparib orally twice a day, with 12 weekly doses of 80 mg/m
2 intravenous paclitaxel, and carboplatin administered every 3 weeks, for 4 cycles [
81,
82,
83]. Patients with a germline
BRCA mutation were then allocated to group II and administered cyclophosphamide and doxorubicin every 2–3 weeks for 4 rounds [
82]. Effective clinical outcomes were observed to be higher in 53% of patients with combined therapies in comparison to patients who received paclitaxel alone (31%). No significant toxicity was observed against Veliparib. [
81,
82,
83].
2.5. Talazoparib (BMN-673)
Talazoparib is a PARP inhibitor that is hypothesized to have a higher effectiveness than olaparib due to the process of PARP trapping, in which a PARP molecule is trapped on the DNA, inhibiting cell division [
84]. Talazoparib is a dual-mechanism PARP inhibitor that traps PARP on DNA [
84,
85]. The phase II study ABRAZO evaluated the efficacy of talazoparib on inpatients with germline
BRCA1/2 mutations before being treated with platinum or multiple regimens [
84,
85]. Clinical efficacy was evaluated in TNBC/HR+ patients at 26%/29%; adverse effects were observed such as neutropenia, thrombocytopenia, anemia, fatigue, nausea, and diarrhea [
85]. A phase III clinical trial was performed to compare the efficacy and safety of talazoparib in TNBC patients [
84,
85,
86]. Clinical efficacy was observed—median PFS was 8.6 months for talazoparib, with a 46% reduction in the tumor, and 5.6 months for chemotherapies such as capecitabine, eribulin, gemcitabine, or vinorelbine [
84,
85,
86]. All key secondary efficacy endpoints (OS, ORR, clinical benefit rate at 24 weeks) demonstrated benefits with talazoparib [
85,
86]. The PARP inhibitor was generally well tolerated, with minimal non-hematologic toxicity and few adverse events associated with treatment discontinuations [
84,
85,
86]. Patients were treated with an anthracycline, with or without taxane as a neoadjuvant [
84]; its primary clinical efficacy was examined, with PFS performed according to RECIST 1.1 criteria: median PFS was 8.6 and 5.6 months in the talazoparib and chemotherapy arms, respectively (HR 0.54; 95% CI: 0.41, 0.71;
p < 0.0001) [
85,
86]. Its clinical approval was considered in EMBRACA (NCT01945775), an open label trial randomizing 431 patients (2:1) who were g
BRCAm
HER2-negative to treatment with talazoparib (1 mg) with no more than 3 prior cytotoxic chemotherapy treatments for metastatic disease. Talazoparib was approved by the FDA for germline
BRCA-mutated (gBRCAm),
HER2-negative locally advanced or metastatic breast cancer. The FDA also approved the BRAC Analysis CDx test for identifying patients with breast cancer with deleterious or suspected deleterious g
BRCAm who are eligible for talazoparib [
85].
2.6. Rucaparib
Rucaparib is an effective inhibitor of PARP1, PARP-2, and PARP-3 in
BRCA-mutated patients (germline and/or somatic). Rucaparib was also found to be effective in HR-deficient patients [
87]. Rucaparib is indicated as a monotherapy treatment for adults who are platinum-sensitive, patients who have been treated with two or more prior lines of platinum-based chemotherapy, and for those who are unable to tolerate further platinum-based chemotherapy [
88]. A multicenter phase clinical trial was performed to establish
BRCA1/2 mutations and earlier treatment with rucaparib. Intravenous, and subsequently oral, rucaparib were evaluated with different dose concentrations [
89]. Efficacy and safety levels were evaluated, such as pharmacodynamics, pharmacokinetic dose-limiting toxic effects, and tolerability [
90]. Intravenous rucaparib was given and the objective response rate was analyzed: 41% of patients showed an ongoing response for at least 12 weeks [
91]. The efficacy and safety of rucaparib in patients with
HER2-negative metastatic breast cancer were associated with
BRCAness phenotype and/or a somatic
BRCA mutations [
87,
88,
89,
90,
91]. Patients received 600 mg rucaparib orally for 21 days or up to the development of the disease. The main endpoint was the clinical benefit rate and secondary endpoints, including PFS, overall survival, safety, and the prognostic value of the
BRCAness signature [
87,
88,
89,
90,
91]. An additional study determined the quantity of sporadic TNBC patients likely to benefit from rucaparib treatment [
87,
88,
89,
90,
91].
2.7. Checkpoint Inhibitors
TNBC is pushing to improve treatment by answering questions regarding biomarkers of response, defining the utility of neoadjuvant approaches, and exploring potential combinations of checkpoint inhibitors and PARP inhibitors. The FDA approved the nab-paclitaxel (Abraxane) with atezolizumab (Tecentriq) for patients with metastatic PD-L1-positive TNBC. The approval was based on the phase 3 IMpassion130 trial (NCT02425891), which established a 38% decrease in the risk of disease development with the combination vs. placebo plus nab-paclitaxel in this patient population. Pembrolizumab is a second approved checkpoint inhibitor drug, approved by the FDA in Nov 2020, for patients with metastatic TNBC whose tumors express a PD-L1 combined positive score (CPS) of 10 or higher, as determined by an FDA-approved test. Pembrolizumab also demonstrated proof of concept as a neoadjuvant based on findings from the phase II I-SPY2 trial (NCT01042379); pembrolizumab neoadjuvant plus chemotherapy extended pathologic complete response (pCR) rates by 13.6 percentage points compared with chemotherapy alone for patients with early TNBC (95% CI, 5.4–21.8; p < 0.001).
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines9111512