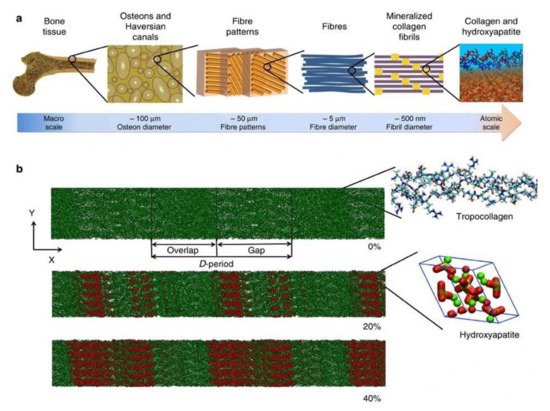
Biomineralization diversifies the mechanical properties of various connective tissues including bone. For instance, mineralization is absent in type 1 collagen of dermis to maintain skin texture, while it is highly mineralized in bone and teeth to form rigid structures [
65]. Mineralization of bone follows a two-step process. Initial step involves the formation of HAP inside the EVs and followed by the propagation into the extracellular matrix. The EVs are typically 50-200 nm in diameter which are produced from the plasma membrane of osteoblasts, chondroblasts and odontoblasts [
33]. The mechanism behind the release of EVs forms these cells is not yet known. Interestingly, the EVs differ in their membrane composition compared to the originating cell type, marked with the presence of several phospholipids, specifically phosphatidylserine which shows high affinity for calcium ions [
66,
67,
68,
69]. EVs are also rich in annexins A5, A2, A6 and calbindin D9k, ALP, carbonic anhydrase, collagen x, type III sodium-phosphate transporter, Phosphatase, Orphan 1 (PHOSPHO1), and nucleotide pyrophosphate phosphodiesterase [
70,
71,
72,
73]. Calcium binding proteins, phospholipids, and bone sialoprotein (BSP) mediate calcium accumulation in EVs [
74]. Incorporation of calcium in EVs is facilitated by the formation of calcium channels by membrane bound annexins. The role of various annexins and matrix vesicles in bone mineralization is discussed in depth and more insights can be acquired by reading Ansari et al. [
75]. Next, phosphates are provided by type III Na/P
i cotransporter present on both EVs membrane and cell membrane [
76,
77]. In addition to this, PHOSPHO1, a cytosolic phosphatase also contributes to phosphate levels by catalyzing the hydrolysis of phosphocholine and phosphoethanolamine [
78,
79]. When the accumulation of both calcium and phosphate exceeds solubility point for calcium phosphate it leads to the deposition as hydroxyapatite inside the EVs. In the next step of mineralization, hydroxyapatite crystals penetrate EVs membrane to reach the extracellular space and undergo crystal elongation. Elongation is highly dependent on extracellular concentrations of calcium and phosphate ions outside the EVs [
73,
75]. Sufficient levels of calcium and P
i support the formation of new apatite crystals, which propagate in clusters around the EVs and fill in between collagen fibrils in the ECM in a cross-fibrillar pattern [
19]. The mechanism of mineralization is diagrammatically represented in
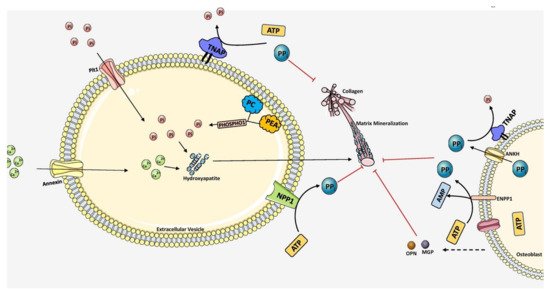
Figure 3.
Figure 3. Overall scheme of matrix mineralization mediated by extracellular vesicles (EV) released from osteoblasts. The growth of hydroxyapatite inside EVs is mediated by the PHOSPHO1 which supplies P
i by hydrolyzing PC and PEA in the plasma membrane. P
it1 transporter also pumps P
i into the EVs. Calcium is transported inside by Annexin (A1, A2, A4, A5, A6, A7) channels. Together, hydroxyapatite crystals are formed inside the EV. It then penetrates through the vesicle membrane and elongates extracellularly utilizing P
i and Ca
2+ ions. PP
i inhibits mineralization which is produced by ATP hydrolysis mediated by NPP1. TNAP hydrolyzes PPi into P
i, essential for hydroxyapatite growth. Extracellular PP
i is also provided by ANKH and ENPP1 situated in the plasma membrane of osteoblasts. Mineralization is regulated tightly by maintaining the balance between PP
i and P
i ratio. The components in this figure were modified from Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License. (
http://smart.servier.com, accessed on 1 August 2021).
Propagation around clusters is mediated cooperatively by EVs and adjacent collagen molecules. Since mineralization is central to bone, our body prevents ectopic biomineralization by various ingenious inhibitors. In bone, to eliminate the inhibitors of biomineralization especially PP
i, osteoblasts express TNAP to annul its inhibitory effect via hydrolysis and generate phosphate ions promoting mineralization. TNAP not only provides phosphate ions for mineralization it also promotes the formation of HAP nanocrystals in the collagen matrix of bone by destabilizing the amorphous phase of the mineral. Also, local inhibitors of biomineralization attenuates pathological mineralization in soft tissues such as cardiac arteries, valves and hard/soft tissue interfaces such as tendon-bone attachments, ligament-bone attachments and cranial sutures. It is noteworthy to mention that, the inorganic phosphate (P
i) to pyrophosphate (PP
i) ratio is crucial for mineralization as PP
i inhibits hydroxyapatite formation [
80]. ANKH, a human homolog of ank gene product, is essential for the extracellular transportation of cytosolic PP
i [
81,
82]. An increase in PP
i/P
i ratio will inhibit spontaneous precipitation of HAP. The PP
i concentration is rightly maintained in forming bone by osteoblasts and acts as a repository of compartmentalized phosphate till it is hydrolyzed [
80]. Extracellular polyphosphate (polyP
i) also participates in phosphates transport and compartmentalization. Poly-P
i granules chelate calcium ions to form neutrally charged amorphous complexes [
83,
84]. Fetuin A is a systemic inhibitor of mineralization which prevents growth of nascent crystal nuclei in blood and facilitates its recycling by macrophages [
85,
86,
87,
88]. One fetuin is known to sequester 54–72 phosphate ions and 90–120 calcium ions. Fetuin A possesses strong affinity to bone and constitutes 25% of the non-collagenous proteins in bone [
89]. It binds with both Ca and PO
42− ions to form calciprotein particles about 30–150 nm in size [
90]. TNAP, expressed highly in osteoblasts, is very essential for the second step of mineralization by decreasing the levels of PP
i and providing P
i for hydroxyapatite formation. Therefore, in bone, mineralization is a well-coordinated, cell mediated dynamic process allowing the exchange of phosphate and calcium ions. In soft tissues, mineralization is generally inhibited by high PP
i/P
i ratio. The polyP
i granules are also known to contain alkaline phosphatases and when activated, hydroxyapatite crystals nucleate inside the granules displacing protein components to the surface forming a crystalline core surrounded by an amorphous shell [
84]. Skeletal tissue biomineralization is also promoted by bone sialoprotein (BSP), i.e., a calcium-binding small integrin-binding ligand N-linked glycoprotein (SIBLING) aids in nucleation of hydroxyapatite mineral [
91]. It is only present in the ECM of mineralized tissues such as bone and teeth. Another promoter of ECM mineralization, Dentin matrix acidic phosphoprotein1 (DMP1) is involved in the stabilization of disordered mineral precursors and directs them to collagen fibrils [
92,
93]. DMP1 binds with mineral forming mineral-protein complexes facilitating electrostatic interaction with collagen fibrils during mineralization. Patients with DMP1 gene deletions or mutations are characterized by osteomalacia with autosomal recessive hypophosphatemic rickets [
94]. OPN and matrix Gla protein (MGP) are mineralization inhibiting proteins which regulate mineral growth in bone and dentition to adjust mineralization or involve complete inhibition of mineralization [
95,
96]. Nascent mineralization foci are associated with osteopontin (OPN) and found widely in the bone matrix. For instance, OPN deficient mice exhibit a hypermineralized skeleton postnatally starting at 12 weeks [
97]. OPN plays a crucial role as interfacial protein in adjoining new bone to old bone and also to implant surfaces following bone graft surgeries. It is actively found in sites where mineralization should be abolished abruptly such as periodontal ligament, and entheses [
98]. OPN knock-out mice failed to curb crystal growth with increased size of mineral crystals but do not show any visible skeletal changes [
99].
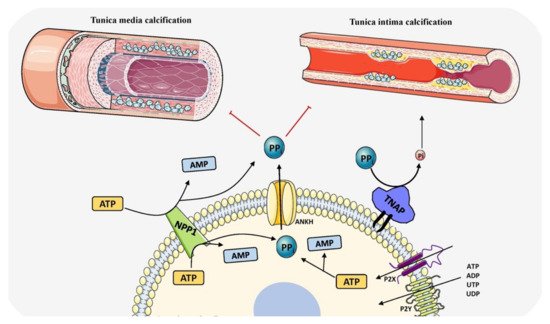
Despite the precise inhibition of mineralization at soft-tissue sites, arteries and articular cartilage are susceptible for ectopic mineralization which leads to detrimental effects. Therefore, mineralization inhibition requires strict regulation involving additional elements. Vascular smooth muscle cells and chondrocytes orchestrate this inhibition by secreting Matrix Gla protein (MGP) into their extracellular matrix (ECM) [
100]. Singleton Merten syndrome and Keutel syndrome associated with defects in MGP gene results in rupture of the aorta and premature fusion of growth plates in the long bone due to pathological mineralization [
101]. Other mineralization inhibitors such as aspartic acid-rich motif (ASARM) and matrix extracellular phosphoglycoprotein (MEPE) also fine tune mineralization [
102,
103]. MEPE knock out mice show increased bone mass and trabecular density but abnormalities in cancellous bone [
104]. MEPE along with dentin matrix protein (DMP1) and Phosphate-regulating neutral endopeptidase (PHEX) regulates mineralization, phosphate levels and turnover of bone by affecting fibroblast growth factor (FGF)-23 expression [
105]. Imbalance in mineralization promotion and inhibition at required areas in the body leads to various pathologies. The following sections, we will discuss the pivotal role of TNAP in various organs with respect to mineralization.