The mechanistic target of rapamycin (mTOR) pathway has manifold functions in the liver involving both the regulation of metabolic activities in healthy organs and processes in the progression of diseases and tissue regeneration. To perform all these functions, the serine/threonine protein kinase mTOR forms multi-protein complexes termed mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), which have different compositions and responses to upstream signals [1].
- mTOR
- Hedgehog
- liver
- hepatocytes
- cyclopamine
- rapamycin
- mTOR/Hedgehog interaction
- primary mouse hepatocytes
-
Definition
The mechanistic target of rapamycin (mTOR) pathway has manifold functions in the liver involving both the regulation of metabolic activities in healthy organs and processes in the progression of diseases and tissue regeneration. To perform all these functions, the serine/threonine protein kinase mTOR forms multi-protein complexes termed mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), which have different compositions and responses to upstream signals [1].
-
Introduction or History
In the liver, mTORC1, which consists of mTOR, mammalian lethal with Sec13 protein 8 (mLST8), Dishevelled, Egl-10 and Pleckstrin domain-containing mTOR-interacting protein (Deptor), regulatory-associated protein of mTOR (Raptor) and proline-rich protein kinase B (Akt) substrate (Pras40), is critical for controlling metabolic processes. Therefore, the main role for mTORC1 is the regulation of the energy status, which it mediates by promoting lipogenesis and lysosome biogenesis as well as inhibiting lipophagy and autophagy in response to growth factors, amino acids and oxygen levels [2]. In this process, mTORC1 regulates mitochondrial oxidative metabolism by initiating the translation of important mitochondrial genes located in the nucleus via Eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) downstream signalling [3,4]. However, Ribosomal protein S6 kinase beta-1 (p70S6)-induced downstream signalling can also influence mitochondrial ATP production and energy metabolism [5]. Similar to mTORC1, mTORC2 consists of mTOR, mLST8 and Deptor, and it also contains rapamycin-insensitive companion of mTOR (RICTOR) and mammalian stress-activated protein kinase-interacting protein (mSIN1) [2]. In contrast to mTORC1, mTORC2 is responsible for cell survival and migration, and it is closely associated with the insulin/phosphoinositide 3-kinase (PI3K) pathway and Akt signalling pathway [1]. In this regard, recent studies have shown that mice in which mTORC2 is downregulated in the liver (via deletion of RICTOR) cannot sense satiety while in a fed state because of enhanced gluconeogenesis and inhibited glycolysis, glycogen synthesis, and lipogenesis [6]. In accordance with their different agonists in vivo, both complexes have different sensitivities to inhibitors, such as rapamycin, which mainly inhibits mTORC1 downstream signalling via p70S6 [7], whereas TORIN supresses both mTORC1 and mTORC2 signalling [8].
Although the molecular mechanisms driving the regulation of metabolic processes via mTOR signalling are well understood in principle, there are still major gaps in the research on the crosstalk of mTOR with signalling pathways known to serve as gatekeepers for lipid metabolism in the liver. Recently, we discovered that the Hedgehog (Hh) pathway is an essential player in liver lipid metabolism and metabolic zonation, influencing circadian rhythms [9,10,11]. The Hh pathway can be induced by binding of the Hh ligands Indian Hh, Sonic Hh and Dessert Hh to the receptors Patched (PTCH). Ligand binding activates the co-receptor Smoothened (SMO), which results in the release of the full-length glioma-associated oncogene family zinc finger (GLI) transcription factors 1–3 from the suppressor of fused (SUFU), with which it had been sequestered. Finally, the GLI transcription factors are translocated to the nucleus to activate Hh-specific target genes [12].
With respect to the crosstalk of the mTOR and the Hh pathway, oncological contexts, such as basal cell carcinoma and gastrointestinal cancer, including oesophageal cancers, are particularly well known and described [13,14]. However, the conditions in tumours characterized by a strong activation of the Hh and/or mTOR signalling cascades are completely different than those in healthy tissue. To date, no investigations have been carried out to determine the crosstalk between the Hh and mTOR pathways in healthy liver and to indicate the ways they may co-modulate aspects of hepatic metabolism. However, since we consider it immensely important to understand the interactions of the networks formed by these highly active metabolic pathways, we conducted this study. Since energy metabolism is known to be affected by liver sexual dimorphism, we also wanted to know whether the crosstalk differs in males and females. By using mice in which hepatocyte-specific Hh has been deleted and in vitro experiments, we demonstrate a strong mutual interaction between Hh signalling and the mTOR pathway in healthy mouse hepatocytes.
-
Data, Model, Applications and Influences
Hh/mTOR Crosstalk in Hepatocytes
Currently the interaction of Hh and mTOR signalling is mainly examined in cancerous tissues. The dysregulation of both pathways plays a role in the development and progression of a variety of cancers, and therefore, Hh and mTOR are targets for clinical treatments. However, the number of tumour types in which Hh and mTOR interact is as large as the variability of their postulated mutually interactions. Some studies have shown that mTOR signalling can directly and indirectly regulate the activation of Gli factors, stimulating non-canonical Hh signalling [13,29]. Others have suggested that mTOR signalling is required for Hh signalling [30,31]. In contrast to the mechanisms described for different cancer types, our female mice with hepatocyte-specific Hh KO exhibit reduced mTOR activity, as was shown through reduced ATP production and mTOR-related gene expression. In addition, low levels of phosphorylated mTOR were found in the male SAC-KO mouse hepatocytes and accumulated p62 was detected in the hepatocytes from both sexes. The accumulation of p62 is a sign for impaired autophagy and additionally is related to chronic liver diseases [32]. Furthermore, it is well known that the mTOR pathway regulates autophagy [2]; therefore, these findings indicate that the absence of Hh signalling can regulate mTOR activity. In contrast, in their 2008 study, Parathath et al. described Hh activation via SHH-induced inhibition of mTOR/p70S6 signalling [33]. However, in the current study, the Akt signalling phosphorylation array analysis revealed increased phosphorylation of the two mTORC1 downstream molecules p70S6 and 4E-BP1 in the male SAC-KO mouse hepatocytes. According to this finding, the IPA software predicted the activation of the mitochondrial transport chain and the mTOR pathway in the male SAC-KO mice, which confirmed the results from Parathath et al. 2008 [33], although the ratio of pmTOR-to-mTOR was reduced. In contrast, the phosphorylation of Akt was reduced and the phosphorylation of RSK1 and RSK2 was elevated. Both findings indicate the inhibition of mTOR signalling [34].While cyclopamine binds to the receptor SMO and thus inhibits the Hh pathway in hepatocytes at the same position where it was blocked in the SAC-KO mice, it did not have the same effect in the short time of 24 h in vitro on hepatocytes from C57BL/6N mice. In the SAC mice, the Hh pathway is blocked in the hepatocytes 9 days after fertilization [18]. Therefore, the hepatocytes lacked Hh signalling for 3 months before they were analysed. During this time these hepatocytes were embedded in their natural environment prior to analyses. In contrast, the Hh signalling in the hepatocytes from the C57BL/6N mice was blocked for only 24 h after preparation in the 2D cell culture; therefore, they were exposed to impaired Hh signalling for a much shorter time and were not exposed to systemic effects. Unfortunately, we could not detect significant downregulation of Gli1 and Ptch1 in primary hepatocytes treated with cyclopamine for 24 h as it is known for numerous other cell types [35,36]. Several reasons for this apparent discrepancy are discussed below. Regarding mTOR signalling, in combination with rapamycin, cyclopamine had an even stronger effect on mTOR signalling compared to rapamycin alone. This synergistic effect was also observed in the 2015 study by Zuo et al. using a xenograft mouse model for biliary tract cancer treated with a combination of vismodegib, which is also a SMO inhibitor, and rapamycin [37]. However, our results revealed an interaction of Hh and mTOR signalling in healthy liver hepatocytes. Interestingly, the effect of the treatment of the primary hepatocytes with Torin exceeded the impact of cyclopamine in combination with rapamycin on mTOR signalling in most cases; however, the addition of cyclopamine to Torin had no added effect. In contrast to rapamycin, Torin inhibits the complete downstream signalling pathways of mTORC1 and mTORC2. Because the Torin-induced inhibition was not enhanced by adding cyclopamine, Torin may block the point where these pathways interact. mTORC2 is only indirectly affected by rapamycin via a feedback mechanism involving p70S6 and Akt. We suggest that Hh has an activating effect on mTORC2, which attenuates the inhibitory effect of rapamycin on mTOR signalling through the Akt feedback mechanism (Figure 7). This idea is supported by the results from a 2008 by Ikenoue et al., which showed that the mTORC2-mediated phosphorylation of Akt was required for the maximum level of Akt-activated signalling to be induced [38]. Therefore, cyclopamine seems to prevent Hh from activating mTORC2, revealing the full strength of the inhibition through rapamycin. However, so far, we cannot definitively exclude possible off-target effects of cyclopamine or the involvement of the non-canonical Hh pathway. Therefore, further studies are needed to clarify these issues.
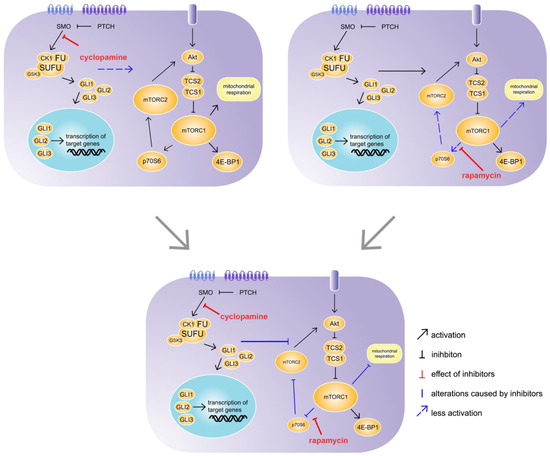
Figure 7. Synergistic effect of cyclopamine and rapamycin. C57BL/6N hepatocytes were treated with cyclopamine and/or rapamycin for 24 h. The treatment with cyclopamine had no effect on mechanistic target of rapamycin (mTOR) signalling, whereas the treatment with rapamycin had weak inhibitory effects on p70S6 downstream signalling and mitochondrial respiration. However, the simultaneous treatment with cyclopamine and rapamycin exhibited strong inhibitory effects on p70S6 downstream signalling as well as mitochondrial respiration, revealing an impact of Hh signalling on the mTOR pathway. SMO: Smoothened, PTCH: Patched, CK1: Casein kinase 1, FU: Fused, SUFU: Suppressor of Fused, GSK3: Glycogen synthase kinase 3, GLI1-3: glioma-associated oncogene family zinc finger 1-3, Akt: Protein kinase B, TCS: Tuberous sclerosis complex, mTORC1/2; mechanistic target of rapamycin complex 1/2, 4E-BP1: Eukaryotic translation initiation factor 4E-binding protein 1, p70S6: Ribosomal protein S6 kinase beta-1.
Hh/mTOR Crosstalk Influences Mitochondrial Metabolism and Therefore the Energy State of the Cell
It is well known that the mTOR pathway regulates mitochondrial metabolism. The downstream signalling of mTORC1 influences mitochondrial biogenesis, morphology and ATP production capacity [3,39,40]. In contrast, the indications to date suggest that the Hh pathway is not closely connected with mitochondrial metabolism. Interestingly, hepatocytes from the female SAC-KO mice showed suggestive evidence for significantly reduced ATP production, which was consistent with the prediction of the IPA software based on proteome data, thereby linking repressed Hh signalling with mitochondrial function. Our results were partly consistent with a 2017 study from Yao et al. showing that Hh activation based on SHH led to an increase in mitochondrial mass, elongation, membrane potential and respiratory activity in hippocampal neurons [41]. However, we did not detect any changes in the mitochondrial mass of the hepatocytes from the SAC-KO mice. Our results were consistent with those from the study by Kalim et al., who showed that mTOR-related changes in mitochondrial mass were dependent on Raptor [42], which was unchanged in our SAC hepatocytes. In addition, the hepatocytes from the SAC-KO mice exhibited increased mitochondrial membrane potential, which also is an opposite finding than that of the Yao et al. study in 2017 [41]. However, it is known that inhibition of ATP synthase can lead to increased mitochondrial membrane potential [43]. Therefore, our results suggest that repressed Hh signalling leads to the inhibition of ATP synthase, a supposition that should be examined in further studies. Moreover, we detected elevated mRNA levels of mitochondrial carrier proteins involved in the citric acid cycle in the hepatocytes from the male but not from the female SAC-KO mice, although male mouse hepatocytes showed no alteration in ATP production. In the female SAC-KO mouse hepatocytes, we detected an increase in the gene expression of the adenine nucleotide translocator Slc25a4. This upregulation could have been a response to the reduced ATP levels and correlates with the striking increase in the proton leak [44]. Additionally, it is known that the proton leak depends on the mitochondrial membrane potential [43]. Therefore, the hyperpolarization of the mitochondria may have also contributed to the increased proton leak. However, the question remains: how does Hh signalling regulate mitochondrial metabolism? One possible explanation may involve the direct interaction of Hh with mTOR signalling. This idea is supported by the synergistic effect of rapamycin and cyclopamine on the inhibition of the maximal respiration and the ATP production rates of the hepatocytes. As mTOR is the main regulator for mitochondrial metabolism, the synergistic inhibition of mTOR signalling through cyclopamine and rapamycin affects the oxidative phosphorylation in mitochondria. Nevertheless, it is possible that cyclopamine influences oxidative phosphorylation independently from the canonical Hh signalling pathway, as it was postulated by Kalainayakan et al. in 2019 [45]. This assumption is supported by the fact that no significant changes were observed in the gene expression of Gli1 and Ptch1 after exposure to cyclopamine. However, the reasons for this observation may be manifold. Firstly, as reported by Ozretić et al. 2017 in tumour cells, the strength of the inhibitory effect of cyclopamine on the mRNA levels of Gli1 and Ptch1 may depend strongly on the cell type [46]. Moreover, specifically in hepatocytes, we recently found that Gli1 is part of a Gli-network and behaves different than in other cell types [25]. Furthermore, it is known that the expression of both genes is subject to pronounced diurnal oscillation [11]. Thus, measurements at a single time point may not be the best choice for estimating Hh pathway activity. In addition, it would be interesting to investigate possible off-target effects of cyclopamine. Therefore, further studies are needed to reveal the exact molecular mechanism of the impact of Hh signalling on mitochondrial metabolism.
This entry is adapted from the peer-reviewed paper 10.3390/cells9081817