Ghrelin is an endogenous ligand for the ghrelin receptor, previously known as the growth hormone secretagogue receptor. This hormone is mainly produced by endocrine cells present in the gastric mucosa. The ghrelin-producing cells are also present in other organs of the body, mainly in the digestive system, but in much smaller amount. Ghrelin exhibits a broad spectrum of physiological effects, such as stimulation of growth hormone secretion, gastric secretion, gastrointestinal motility, and food intake, as well as regulation of glucose homeostasis and bone formation, and inhibition of inflammatory processes.
- ghrelin
- anti-inflammatory effects
- pretreatment
- protection
- healing
1. Ghrelin and Its Synthesis
Ghrelin, a 28-amino acid peptide, was primary isolated by Kojima et al. from rat and human stomachs in 1999 [1][2][3]. The main source of endogenous ghrelin in the body is the stomach [1][4]. Ghrelin is created from its 117-amino acid precursor, preproghrelin, which consists of a 23-amino acid signal sequence and the 94-amino acid proghrelin [1][5]. The proghrelin is further converted into acyl-ghrelin, des-acyl ghrelin, and obestatin [5][6][7].
Most studies show that the majority of ghrelin in circulation exists in the form of des-acyl ghrelin [8][9][10]. On the other hand, Blatnik et al. [11] postulate that these observations are a result of errors in sampling, handling, collection, and assessment of serum ghrelin. Blatnik et al. analyzed the acyl ghrelin plasma stability by LC-MS/MS and revealed that acyl ghrelin is enzymatically and chemically converted to des-acyl ghrelin in the presence of active serine proteases and HCl. They concluded that that normally all circulating ghrelin is acylated, and des-acyl ghrelin should not be detectible in healthy human plasma under optimal sample handling and assaying conditions [11].
Acyl-ghrelin is considered to be an active form of this hormone [6][8][12]. Acylation is necessary to stimulate the growth hormone secretagogue receptor (GHSR-1a), currently known as the ghrelin receptor [13]. The ghrelin receptors are mainly expressed in the pituitary gland and hypothalamus, but were also present in other tissues and organs [5][13][14][15]. Expression of ghrelin receptor is highly sensitive to the level of growth hormone. In growth hormone-deficient animals, expression of mRNA for ghrelin receptor is increased. On the other hand, an increase in serum growth hormone level reduces the expression of ghrelin receptor [16].
Acylation of ghrelin is catalyzed by the ghrelin O-acyltransferase (GOAT), which was discovered in 2008 [17]. GOAT belongs to a family of hydrophobic membrane-bound acyltransferases [17][18]. Des-acyl ghrelin does not bind to ghrelin receptor, GHSR-1a, and is deprived of growth hormone releasing activity. However, this form of ghrelin may exhibit some non-endocrinological activity, such as the protection of endothelial cells and cardiomyocytes in the heart, regulation of food intake, gastric and pancreatic secretion, gut motility, adipogenesis, stimulation of bone formation, insulin secretion, and prevention of skeletal muscle atrophy [2][3][19][20].
Acyl-ghrelin acting on ghrelin receptor (previously known as GHSR-1a) strongly and dose-dependently stimulates synthesis and release of growth hormone in the anterior lobe of the pituitary gland [1][3]. This effect of ghrelin is mainly related to direct stimulation of somatotropes. However, ghrelin also stimulates the liberation of growth hormone via an indirect pathway. Ghrelin, acting on neurons expressing growth hormone-releasing hormone (GH-RH) in the hypothalamus, leads to the secretion of GH-RH by these neurons. Subsequently, GH-RH reaches somatotropes in the anterior part of the pituitary and stimulates them to release the growth hormone [21]. The ghrelin receptor is a G-protein-coupled receptor and signals via a Gq/11 alpha-subunit, that results in the activation of phospholipase C and the synthesis of inositol triphosphate (IP3), and releases Ca2+ from the endoplasmic reticulum [12][22]. On the other hand, Ge et al. [23] have reported that stimulatory effect of ghrelin on ghrelin receptor can be reduced by liver-expressed antimicrobial peptide 2 (LEAP2), an endogenous antagonist of ghrelin receptor. LEAP2 is produced in the liver and small intestine. This peptide inhibits ghrelin receptor activation by ghrelin, leading to reduction in the major effects of ghrelin in the body, such as food intake, growth hormone release, and maintenance of viable glucose levels during fasting. Secretion of endogenous LEAP2 is suppressed by food restriction, and this effect leads to increased reactivity of ghrelin receptor to the action of ghrelin [23]. Moreover, studies performed on neoplastic cell lines suggest that ghrelin may activate P13K/GTP-Rac [24], GHSR/P13K/Akt [25], and GHSR/CaMKII/AMPK/NFκB [26] signaling pathways.
Apart from ghrelin receptor, there is another type of growth hormone secretagogue receptor, GHSR-1b, but this receptor seems to be not biologically active. Its role is unknown [3].
Ghrelin is mainly synthesized in the gastric oxyntic mucosa, but its presence was also found in the oral cavity, small and large bowel, pancreas, thyroid, lung, testis, myocardium, kidney, brain cortex, brain stem, pituitary, hypothalamus, and immune cells [14][15][27][28]. In rats and dogs, ghrelin is produced in the stomach by the neuroendocrine X/A-like cells [29][30]. These cells are small and round. They have no contact with a stomach lumen. In the human stomach, ghrelin is produced in endocrine cells called P/D1 cells. In the small and large bowel, there are two types of ghrelin-secreting cells: closed-type cells with triangular and elongated shapes, and opened-type cells with their apical cytoplasmic process contacting to the intestinal lumen [1][30]. In the pancreas, ghrelin is produced by endocrine and exocrine cells [15][31][32].
In the case of a decrease in the production of ghrelin in the gastric mucosa, a compensatory increase in the production of this peptide in other areas of the body may occur. Partial resection of gastric mucosa, as a result of bariatric surgery leads to a decrease in serum ghrelin level in the early postoperative period [33]. Later, however, this level returns to the initial value [33] or may be even higher than before the operation [34]. In line with those observation are findings of animal studies performed by Camacho-Ramirez et al. [35], who found that a severe reduction in gastric secretion of ghrelin leads to an increase in the islet ghrelin-secreting epsilon cell population, leading to a subsequent recovery of basal serum ghrelin levels.
2. Physiological Action of Ghrelin
The main physiological function of ghrelin is dose-dependent stimulation of growth hormone release from the pituitary gland [1][2]. The endocrine effects of ghrelin also include the stimulation of prolactin, cortisol, and adrenocorticotropic hormone secretion [36][37].
Ghrelin is responsible for a positive energy balance. This hormone increases food intake and fat deposition [2][38][39]. The increase in appetite, known as orexigenic effect, is mediated by stimulation of hypothalamic neurons releasing neuropeptide Y, orexin, and agouti-related protein (AgRP), as well as by inhibition of hypothalamic proopiomelanocortin (POMC) neurons [40][41][42]. Among orexigenic peptides stimulating appetite, ghrelin is the only one acting peripherally, whereas all other orexigenic peptides are acting centrally [12]. Besides the stimulation of food intake, ghrelin promotes carbohydrate oxidation and inhibits fat utilization, leading to positive energy balance [43]. The plasma level of ghrelin is negatively correlated with BMI and food intake. For this reason, the plasma concentration of ghrelin is enhanced by anorexia nervosa, starvation, and cachexia, while obesity leads to the opposite effect [44]. Food intake decreases the plasma ghrelin levels, however the degree of this reduction depends on the type of nutrients present in the food. The strongest effect is observed after protein consumption, smaller in case of carbohydrates, and the smallest after the ingestion of lipids [45] (Figure 1).
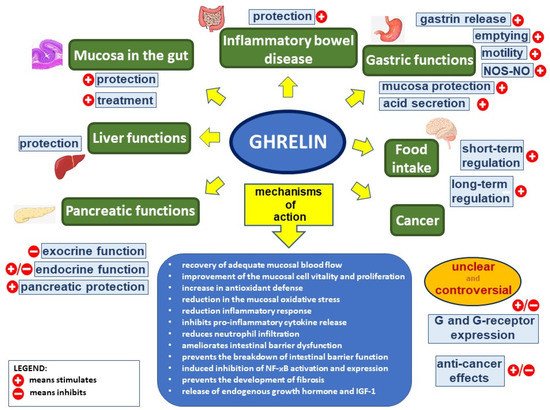
Figure 1. Ghrelin’s effect in the digestive system. Figure legend: NOS–NO—nitric oxide synthase–nitric oxide, G—ghrelin, NF-κB—nuclear factor kappa-light-chain-enhancer of activated B cells, and IGF-1—insulin-like growth factor-1; (+) means stimulates, (−) means inhibits.
Ghrelin stimulates gastric motility and gastric emptying [2][46][47]. Impact of ghrelin on the exocrine secretory activity in the stomach is unclear. Gastric acid secretion is dose-dependently increased by the ghrelin administrated peripherally, through a mechanism involving vagal nerve activity and histamine release [46][48][49][50]. Ghrelin effects on gastric acid secretion are in synergy with effects of gastrin [12][51][52]. On the other hand, ghrelin administrated centrally exhibits the opposite effect, inhibiting gastric acid release [12][53][54].
Circulating ghrelin inhibits pancreatic exocrine secretion. Zhang et al. [55] demonstrated that intravenous administration of ghrelin reduces the 2-deoxy-D-glucose- and cholecystokinin-stimulated pancreatic exocrine secretion in anesthetized rats. Moreover, ghrelin inhibits the potassium-stimulated amylase secretion in isolated pancreatic lobules [2][55]. On the other hand, Sato et al. [56] reported, that intracerebroventricular administration of ghrelin rises pancreatic exocrine secretion in conscious rats, and the mechanism of this effect involves the vagal nerves [2][56]. The effect of ghrelin on pancreatic endocrine secretion was initially unclear. Early studies have shown that ghrelin increases insulin secretion by pancreatic β-cells [44][57][58], while next studies have reported that ghrelin inhibits insulin release in the islets of Langerhans [44][59][60]. Currently, it is commonly accepted that ghrelin inhibits glucose-dependent insulin secretion, acting directly on beta-cells in pancreatic islets [44][61][62]. Physiologically, this mechanism is mainly related to ghrelin expressed in pancreatic islets and released into pancreatic microcirculations. Ghrelin has been shown to inhibit insulin release in mice, rats, and humans. Ghrelin antagonists or genetic blockades of islet-derived ghrelin markedly augment glucose-induced insulin release [63]. Inhibition of glucose-induced insulin secretion by ghrelin involves direct interaction of ghrelin with ghrelin receptor coupled to novel cAMP/TRPM2 (cyclic adenosine monophosphate/transient receptor potential melastatin 2) signaling in β-cells [64]. This β-cell unique ghrelin receptor with insulinostatic signaling largely accounts for the systemic effects of ghrelin on circulating glucose and insulin levels. Activation of ghrelin receptor in β-cells inhibits the glucose-induced cAMP and TRPM2 production, and suppresses the glucose-induced [Ca(2+)](i) increase in the β-cell, leading to inhibition of insulin release by β-cells in pancreatic islets [63][64].
There are other functions of ghrelin that are worth mentioning. Vestergaard et al. demonstrated that acyl-ghrelin infusion increases thirst sensation in humans, without affecting diuresis and renal sodium excretion [65]. Ghrelin has been reported to exhibit antidepressant effects [66]. Moreover, Liu et al. showed that ghrelin promotes neural differentiation of adipose tissue-derived mesenchymal stem cells, through the activation of β-catenin and AKT/mTOR signaling pathways [66][67].
3. Protective, Anti-Inflammatory, and Healing Effects of Ghrelin in the Digestive System
Protective and healing effects of ghrelin were found in all parts of the digestive system, from the oral cavity to the colon. The influence of endogenous ghrelin level on functional gastrointestinal disorders is unclear. There are studies suggesting that functional dyspepsia is associated with the higher level of serum acyl- or des-acyl ghrelin; however, there is a similar number of articles suggesting the opposite relationship between serum level of acyl- or des-acyl ghrelin and incidence of functional dyspepsia [68].
3.1. The Oral Cavity
Ghrelin is synthesized and released by parotid and submandibular salivary glands. Its presence was found in the cytoplasm of striated, intercalated, and excretory ducts, as well as in serous acini of these glands [69]. Ghrelin is also produced and/or present in teeth, taste buds of the tongue, and gingival epithelium, as well as fibroblasts in the lamina propria [70][71][72]. Ghrelin seems to be involved in the tooth development [73]. The concentration of ghrelin in saliva in similar or even higher than that in plasma or serum, the highest level of ghrelin is observed in gingival crevicular fluid [71][74][75]. Proinflammatory factors, such interleukin-1β, increase the expression of mRNA for ghrelin receptor and production of ghrelin receptor in periodontal cells [76]. On the other hand, exogenous ghrelin inhibits the production and release of proinflammatory interleukin-8 by oral epithelial cells stimulated by tumor necrosis factor-α (TNF-α) or lipopolysaccharides [71]. These findings suggest that ghrelin may be involved in endogenous protective mechanisms limiting local inflammation. Moreover, previous experimental studies showed that intraperitoneal administration of exogenous ghrelin significantly accelerate the healing of acetic acid-induced oral ulcers, and that that effect occurs in rats with intact salivary glands, as well in sialoadenectomized rats. The beneficial effect of ghrelin is associated with a reduction in mucosal IL-1β concentration and an improvement of mucosal blood flow, cell vitality, and proliferation. These finding have been confirmed and extended by studies performed on animals [77].
3.2. The Esophagus
Clinical and experimental studies have shown the expression of ghrelin receptor to be increased in Barrett’s mucosa in comparison with normal esophageal squamous epithelium. However, ghrelin administration is without any effect on apoptosis of Barrett adenocarcinoma cell line, OE-19 in vitro. On the other hand, administration of ghrelin seems to inhibit Barrett’s carcinogenesis due to suppression of expression of proinflammatory response [78].
Thomas et al. performed a clinical study concerning the relationship between serum level of ghrelin and Barrett’s esophagus [79]. They found that higher ghrelin concentration is associated with an increased risk of Barrett’s esophagus in comparison to the control population, but not when compared with patients with gastroesophageal reflux disease (GERD). Moreover, they reported that ghrelin concentration is associated with the frequency of GERD symptoms.
On the other hand, there is a group of articles showing the influence of ghrelin administration on the inflammatory response in patients with esophageal cancer treated with esophagectomy. Esophagectomy is a highly invasive procedure leading to extended systemic inflammatory response syndrome (SIRS). Continuous infusion of ghrelin (0.5 μg/kg/h) for 5 days after esophagectomy led to a reduction in SIRS duration and lower C-reactive protein and interleukin-6 levels in comparison to the placebo group. Moreover, ghrelin reduced the incidence of pulmonary complications and the time of the negative nitrogen balance during postoperative period [80]. The effect of continuous infusion of ghrelin for 5 days after esophagectomy led to the better therapeutic effect than intermittent infusion for 10 days [81]. In addition, an early drop in plasma level of ghrelin after esophagectomy may be used as a predictor of prolonged SIRS in postoperative period [82].
Moreover, low level of ghrelin seems to be recognized as a risk factor for the development of esophagogastric junctional and gastric adenocarcinomas [83].
The influence of treatment with ghrelin on the healing of esophageal injury is unknown.
3.3. The Stomach
Gastroprotective effect of ghrelin was shown in different experimental models of gastric ulcers. In 2003, Sibilia et al. showed that central, as well as peripheral administration of ghrelin inhibits the development of ethanol-induced gastric ulcers in rats [84]. The protective effects of ghrelin given centrally were found to be much more pronounced than the effects of ghrelin given peripherally. Pretreatment with nitric oxide synthases inhibitor, N(omega)-nitro-L-arginine methyl ester (L-NAME), or deactivation of sensory nerves by neurotoxic dose of capsaicin abolished the protective effects of ghrelin given centrally. Sibilia et al. concluded that mechanisms of the gastroprotective effects of ghrelin involve nitric oxide (NO) release and activity of sensory nerve fibers [84]. Similar gastroprotective effect of ghrelin in ethanol-induced gastric ulcer was found by Konturek et al. [85]. Intraperitoneal pretreatment with of ghrelin led to dose-dependent inhibition of the development of gastric lesions, and this effect was associated with the improvement of gastric blood flow and reversion of ethanol-induced increase in TNF-α expression in gastric mucosa. Indomethacin administered prior to the induction of ulcers increased gastric mucosa damage and reduced gastroprotective effects of ghrelin [85].
Ghrelin also exhibits a gastroprotective effect in other models of gastric mucosa damage. Pretreatment with ghrelin inhibits the development of gastric ulcers evoked by water immersion and restrain stress (WRS) [86][87], gastric ischemia followed by reperfusion [88][89], intragastric administration of concentrated hydrochloric acid [90] or alendronate [91].
In addition to its protective effect, ghrelin also exhibits the healing effect in the stomach. Administration of ghrelin after induction of gastric ulcers accelerates the healing of gastric ulcers induced by ethanol [92] and acetic acid [93].
3.4. The Small Intestine
Ghrelin has been found to protect the small intestine against damage evoked by ischemia/reperfusion [94][95] and this effect was observed after intracerebroventricular, as well as intravenous, administration of ghrelin. This protective effect was found as reduction in proinflammatory cytokine release and neutrophil infiltration in the intestine and lungs. In addition, ghrelin ameliorated intestinal barrier dysfunction, attenuated intestinal and pulmonary injury, and improved the survival of animals subjected to the gut ischemia/reperfusion-induced damage [94]. Previous studies have also reported that the protective effect of ghrelin against intestinal injury is related to improved intestinal blood flow [95] and promoting the activation of the mTOR/p70S6K signaling pathway [96]. Moreover, intravenous administration of ghrelin receptor antagonist increased the ischemia/reperfusion-induced intestinal and pulmonary injury and animal mortality [94]. This last observation indicates that endogenous ghrelin is involved in maintaining the integrity of the small intestine.
Administration of ghrelin also inhibits the development of experimental damage in the small intestine induced by whole body irradiation [97] and attenuates intestinal barrier dysfunction following intracerebral hemorrhage [98].
Animal experimental studies have also shown that administration of ghrelin exhibits therapeutic effects in injury of the small intestine. Ghrelin accelerates the healing of duodenal ulcers induced by acetic acid [93] or cysteamine [99]. Moreover, ghrelin stimulates intestinal adaptation following a massive resection of the small intestine in parenterally fed rats [100].
3.5. The Liver
Previous clinical studies have shown that a low fasting level of ghrelin is associated with increased risk of developing gallstone disease [101], whereas a high fasting serum level of ghrelin reduces the risk of developing nonalcoholic fatty liver disease (NAFLD) [102]. In line with this last observation are the results obtained by Ezquerro et al. [103]. They found that endogenous ghrelin plays a protective role in NAFLD. An increased acylated/desacyl ghrelin ratio in patients with obesity and NAFLD seems to be related to a compensatory mechanism to overcome TNF-α-induced hepatocyte apoptosis, autophagy, and pyroptosis. The protective effects of ghrelin were also shown in animal models of NAFLD. Nagoya et al. have demonstrated that the fatty liver stimulates the autonomic nervous signal circuits, suppressing the progression of the disease by activating the gastric ghrelin expression and the release of IGF-1 from the liver [104]. Moreover, administration of ghrelin in experimental models of NAFLD was found to exhibit preventive and therapeutic effect in this disease [105]. Administration of ghrelin reduced the NAFLD-induced histological changes in the liver, including necrosis, level of apoptotic cells and inflammation foci. This effect was accompanied by a reduction in serum activity of hepatic enzymes, oxidative stress, and lipid peroxidation markers, as well as a decrease in proinflammatory cytokines level. The protective effect of ghrelin on the liver has been also shown in numerous animal models of liver injury. Treatment with ghrelin reduces the acetaminophen- [106], bile duct ligation- [107], ischemia/reperfusion- [108], and the carbon tetrachloride-induced liver injury [107][109][110]. The above articles have shown that the hepatoprotective effect of ghrelin is associated with its antioxidant, anti-inflammatory, and antifibrotic effects. Moreover, Arıcı and Cetin [110] have shown that administration of ghrelin reduces the carbon tetrachloride-induced coagulation disorders.
3.6. The Pancreas
Ghrelin exhibits a protective and therapeutic effect on the endocrine and exocrine pancreas.
In the endocrine pancreas, acyl- and des-acyl-ghrelin have been also found to promote proliferation and inhibit apoptosis in pancreatic β-cells and human pancreatic islets [111][112]. In addition, des-acyl ghrelin increases islet cell mass and prevents stretozotocin-induced diabetes in newborn rats [113]. A similar protective effect of ghrelin on pancreatic β-cell has been reported by Wang et al. [114]. Exposure of β-cells to palmitate led to a significant increase in β-cells apoptosis. Administration of ghrelin promoted survival and attenuates the palmitate-induced apoptosis in β-cells [114]. An antiapoptotic effect of ghrelin in pancreatic β-cells was also found by other researchers. Diaz-Ganete et al. performed studies on β-cell line and isolated rats’ pancreatic islets. They found ghrelin has no remarkable effect on β-cells in basal condition without presence of noxious factors. However, when β-cells are exposed to proinflammatory cytokines, ghrelin reduces activation of apoptotic mediators and endoplasmic reticulum stress, restores insulin release in response to glucose, and activates cell survival pathways. They suggested that ghrelin could potentially be effective in preventing or slowing the transition from a preclinical to clinical type 1 diabetes by mitigating insulitis-induced β-cell damage [115]. This concept has been supported by further studies performed with animal models of autoimmune diabetes mellitus. Administration of ghrelin before induction of insulitis significantly reduced the development of diabetes, as well as prevented the reduction in the number of β-cells, islet area, islet number, and β-cell proliferation [116].
In the case of the exocrine pancreas, the protective and therapeutic effect of ghrelin is mainly related to the development and course of acute pancreatitis. Animal experimental studies have shown that pretreatment with ghrelin inhibits the development of acute pancreatitis evoked by cerulein [117], pancreatic ischemia with subsequent reperfusion [118] and taurocholate [119][120]. In the case of acute taurocholate-induced pancreatitis, the anti-inflammatory effects of ghrelin were observed not only in the pancreas, but also in the liver and lung [119][120]. The protective effect of ghrelin was also found in cellular models of acute pancreatitis [121][122][123]. On the other hand, inhibition of ghrelin gene expression in pancreatic acinar cells, AR42J cells, results in increased expression of intracellular inflammatory factors after administration of cerulein [124].
As well as the protective effect, exogenous ghrelin was found to exhibit therapeutic effects in experimental acute pancreatitis. Administration of exogenous ghrelin was found to inhibit the inflammatory process and accelerate the recovery in different animal models of this disease, including cerulein- [125] and ischemia/reperfusion-induced acute pancreatitis [126].
There are also clinical studies showing a relationship between endogenous ghrelin and the course of acute pancreatitis in humans. Some reports suggest that serum ghrelin levels may be a prognostic factor in the course of acute pancreatitis. Wang et al. [127] tested serum ghrelin levels in patients with acute pancreatitis. Patients were divided into three groups: patients with (a) mild; (b) moderate severe; and (c) severe acute pancreatitis. On the 1st day of hospitalization, fasting serum ghrelin concentration was significantly lower in patients with pancreatitis in comparison to healthy controls; the serum level of ghrelin also significantly decreased with increasing severity of acute pancreatitis. During the next four days, fasting serum level of ghrelin increased in all groups of patients, but was still lower than in control group. In addition, serum ghrelin was lower in patients with severe acute pancreatitis than in patients with mild or moderate severe acute pancreatitis [127]. A similar initial drop in ghrelin levels with subsequent increase in the course of acute pancreatitis was also found by Panek et al. [128]. Moreover, they concluded that rising serum ghrelin levels in the course of acute pancreatitis may be a marker of recovery and an indicator of the healing process.
On the other hand, there are also articles reporting that ghrelin affects the course of acute pancreatitis and plays an important role in the regulation of inflammatory response [129], but ghrelin serum level is not a useful predictor of the severity of acute pancreatitis [129][130]. The differences in data presented in above-mentioned articles may be the due to the different number of observations, as well as the criteria for collecting the material and methods for determining ghrelin level. However, it should be stated that all of the above articles concerning clinical observation suggest the participation of endogenous ghrelin in anti-inflammatory and regenerative processes in the course of acute pancreatitis.
The role of endogenous ghrelin was also shown in recovery after pancreatic surgery. Sasaki et al. [131] have shown that plasma ghrelin suppression after pancreatoduodenectomy is a useful marker for predicting postoperative complications. This finding is in line with experimental data showing that exogenous ghrelin enhances endocrine and exocrine regeneration of the pancreas after pancreatectomy [132].
3.7. The Large Bowel
The relationship between ghrelin and inflammatory bowel diseases is not clear. Previous studies have shown that patients in the acute phase of Crohn’s disease and ulcerative colitis have higher circulating levels of ghrelin than patients in remission or healthy controls [133][134][135]. In addition, ghrelin mRNA and ghrelin receptor mRNA in colonic mucosa are higher in active IBD patients than in healthy control [136][137]. Moreover, in patients with Crohn’s disease, there is significantly higher percentage of ghrelin-positive peripheral blood T cells than healthy in individuals [136].
There are experimental studies showing protective and healing effect of exogenous ghrelin in colitis. Gonzalez-Rey et al. [138] found that treatment with ghrelin significantly ameliorates the severity of the trinitrobenzene sulfonic acid (TNBS)-induced colitis; as well as colitis evoked by dextran sulfate sodium (DSS). The study was carried out on mice. Administration of ghrelin significantly reduced animals’ weight loss, diarrhea, and inflammation, as well as increased the survival rate of the animals. In line with these findings were clinical and experimental studies performed by Konturek et al. [137]. In the clinical study, they found that patients with ulcerative colitis exhibit a significant upregulation of mRNA for ghrelin and tumor necrosis factor-α (TNF-α) in colonic mucosa in comparison to healthy controls. The ratio of mRNA expression for ghrelin was found to be well-correlated with the severity of inflammation and expression of TNF-α. The animal study showed that treatment with ghrelin accelerates the healing of TNBS-induced colitis in rats, and this effect is accompanied by an increase in inducible nitric oxide synthase mRNA expression and synthesis of cyclooxygenase 2 (COX-2) in the colonic mucosa. These findings suggest that endogenous ghrelin may protect and accelerate the healing of inflamed colonic mucosa, and that ghrelin could be useful in the treatment of ulcerative colitis [137]. The therapeutic effect of ghrelin in TNBS-induced colitis was also shown by Zhang et al. [139].
Similar protective and/or therapeutic effects of ghrelin were also found in other experimental models of inflammatory bowel disease (IBD). Pretreatment [140] or treatment [141][142] with ghrelin reduces the severity of colitis evoked by acetic acid enema and accelerates the healing in this model of IBD. Moreover, Ozturk et al. [143] suggested that protective and therapeutic effects of nesfatin-1 in acetic acid-induced colitis in rats involve activation of ghrelin receptors.
The beneficial effect of ghrelin administration was also shown in dextran sodium sulfate (DSS)-induced colitis in rats [144] and mice [139][145]. In addition, Cheng et al. [145] reported that ghrelin prevented the breakdown of intestinal barrier function in DSS-induced colitis by inhibiting the activation of nuclear factor kappa B (NFκB). This observation is supported by the findings of Zhang et al. [139], which show that the beneficial effect of ghrelin in DSS-induce colitis involves the inhibition of intestinal cell apoptosis.
On the other hand, there are some experimental data suggesting the proinflammatory effect of ghrelin in DSS-induced colitis in mice. De Smet et al. [146] carried out their study in two series. In the first series, they induced colitis in ghrelin(+/+) and ghrelin(−/−) mice. In the second series, they induced colitis in non-inbred Swiss mice by adding 3% dextran sodium sulfate (DSS) to drinking water and dividing the animals into two groups to treated intraperitoneally with saline or ghrelin. De Smet et al. found that the signs of the severity of colitis, such as body weight loss, histological signs of colonic damage, and colonic level of myeloperoxidase activity and interleukin-1β, were significantly less pronounced in ghrelin knockout mice compared to ghrelin(+/+) mice. Moreover, they found that 10 days treatment of non-inbred Swiss mice with exogenous ghrelin enhances the severity of colitis and promotes the release of proinflammatory cytokines in the colon. In conclusion, the authors suggested that endogenous and exogenous ghrelin enhances the colonic manifestations of dextran sodium sulfate-induced colitis in mice [146]. A similar effect was observed by Liu et al. [147]. They compared the severity of DSS-induced colitis in wild mice and ghrelin receptor (−/−) mice. They found that a lack of ghrelin receptor significantly attenuated the severity of DSS-induced colitis. The concept of proinflammatory effects of ghrelin in colitis is also supported by Tian et al. [148]. They reported that knockdown of ghrelin-O-acyltransferase, an enzyme necessary for the production of active, acylated form of ghrelin, attenuates DSS-induce colitis in mice.
The discrepancy between the therapeutic effect of ghrelin in colitis observed by most authors and the harmful effects of ghrelin presented in the last three articles can be explained by the specificity of colitis induced by DSS administered in drinking water. It should be recognized that the severity of colitis most likely depends on the total amount of DSS taken, as well as the amount of DSS taken per unit of body mass. On the other hand, ghrelin increases food [38] and water [65] intake. This most likely causes the amount of DSS ingested to increase. Thus, the greater damage to the colon in animals with active ghrelin receptors, preserved ghrelin production capacity, and receiving exogenous ghrelin is most likely not a result of the damaging effects of ghrelin, but of the increased intake of DSS.
This entry is adapted from the peer-reviewed paper 10.3390/ijms221910571
References
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660.
- Warzecha, Z.; Dembinski, A. Protective and Therapeutic Effects of Ghrelin in the Gut. Curr. Med. Chem. 2012, 19, 118–125.
- Müller, T.D.; Nogueiras, R.; Andermann, M.L.; Andrews, Z.B.; Anker, S.D.; Argente, J.; Batterham, R.L.; Benoit, S.C.; Bowers, C.Y.; Broglio, F.; et al. Ghrelin. Mol. Metab. 2015, 4, 437–460.
- Ariyasu, H.; Takaya, K.; Tagami, T.; Ogawa, Y.; Hosoda, K.; Akamizu, T.; Suda, M.; Koh, T.; Natsui, K.; Toyooka, S.; et al. Stomach is a major source of circulating ghrelin, and feeding state determines plasma ghrelin-like immunoreactivity levels in humans. J. Clin. Endocrinol. Metab. 2001, 86, 4753–4758.
- Kojima, M.; Kangawa, K. Ghrelin: Structure and function. Physiol. Rev. 2005, 85, 495–522.
- Ceranowicz, P.; Warzecha, Z.; Dembinski, A. Peptidyl hormones of endocrine cells origin in the gut—Their discovery and physiological relevance. J. Physiol. Pharmacol. 2015, 66, 11–27.
- Zhang, J.V.; Ren, P.G.; Avsian-Kretchmer, O.; Luo, C.W.; Rauch, R.; Klein, C.; Hsueh, A.J. Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin’s effects on food intake. Science 2005, 310, 996–999.
- Patterson, M.; Murphy, K.G.; Le Roux, C.W.; Ghatei, M.A.; Bloom, S.R. Characterization of ghrelin-like immunoreactivity in human plasma. J. Clin. Endocrinol. Metab. 2005, 90, 2205–2211.
- Yoshimoto, A.; Mori, K.; Sugawara, A.; Mukoyama, M.; Yahata, K.; Suganami, T.; Takaya, K.; Hosoda, H.; Kojima, M.; Kangawa, K.; et al. Plasma ghrelin and desacyl ghrelin concentrations in renal failure. J. Am. Soc. Nephrol. 2002, 13, 2748–2752.
- Ariyasu, H.; Takaya, K.; Hosoda, H.; Iwakura, H.; Ebihara, K.; Mori, K.; Ogawa, Y.; Hosoda, K.; Akamizu, T.; Kojima, M.; et al. Delayed short-term secretory regulation of ghrelin in obese animals: Evidenced by a specific RIA for the active form of ghrelin. Endocrinology 2002, 143, 3341–3350.
- Blatnik, M.; Soderstrom, C.I.; Dysinger, M.; Fraser, S.A. Prandial ghrelin attenuation provides evidence that des-acyl ghrelin may be an artifact of sample handling in human plasma. Bioanalysis 2012, 4, 2447–2455.
- Delporte, C. Structure and Physiological Actions of Ghrelin. Scientifica 2013, 2013, 1–25.
- Davenport, A.P.; Bonner, T.I.; Foord, S.M.; Harmar, A.J.; Neubig, R.R.; Pin, J.P.; Spedding, M.; Kojima, M.; Kangawa, K. International union of pharmacology. LVI. Ghrelin receptor nomenclature, distribution, and function. Pharmacol. Rev. 2005, 57, 541–546.
- Hattori, N.; Saito, T.; Yagyu, T.; Jiang, B.H.; Kitagawa, K.; Inagaki, C. GH, GH receptor, GH secretagogue receptor, and Ghrelin expression in human T cells, B cells, and neutrophils. J. Clin. Endocrinol. Metab. 2001, 86, 4284–4291.
- Gnanapavan, S.; Kola, B.; Bustin, S.A.; Morris, D.G.; McGee, P.; Fairclough, P.; Bhattacharya, S.; Carpenter, R.; Grossman, A.B.; Korbonits, M. The Tissue Distribution of the mRNA of Ghrelin and Subtypes of Its Receptor, GHS-R, in Humans. J. Clin. Endocrinol. Metab. 2002, 87, 2988–2991.
- Bennett, P.A.; Thomas, G.B.; Howard, A.D.; Feighner, S.D.; van der Ploeg, L.H.; Smith, R.G.; Robinson, I.C. Hypothalamic growth hormone secretagogue-receptor (GHS-R) expression is regulated by growth hormone in the rat. Endocrinology 1997, 138, 4552–4557.
- Yang, J.; Brown, M.S.; Liang, G.; Grishin, N.V.; Goldstein, J.L. Identification of the Acyltransferase that Octanoylates Ghrelin, an Appetite-Stimulating Peptide Hormone. Cell 2008, 132, 387–396.
- Zhao, T.J.; Liang, G.; Li, R.L.; Xie, X.; Sleeman, M.W.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Goldstein, J.L.; Brown, M.S. Ghrelin O-acyltransferase (GOAT) is essential for growth hormone-mediated survival of calorie-restricted mice. Proc. Natl. Acad. Sci. USA 2010, 107, 7467–7472.
- Baldanzi, G.; Filigheddu, N.; Cutrupi, S.; Catapano, F.; Bonissoni, S.; Fubini, A.; Malan, D.; Baj, G.; Granata, R.; Broglio, F.; et al. Ghrelin and des-acyl ghrelin inhibit cell death in cardiomyocytes and endothelial cells through ERK1/2 and PI 3-kinase/AKT. J. Cell Biol. 2002, 159, 1029–1037.
- Porporato, P.E.; Filigheddu, N.; Reano, S.; Ferrara, M.; Angelino, E.; Gnocchi, V.F.; Prodam, F.; Ronchi, G.; Fagoonee, S.; Fornaro, M.; et al. Acylated and unacylated ghrelin impair skeletal muscle atrophy in mice. J. Clin. Investig. 2013, 123, 611–622.
- Tannenbaum, G.S.; Lapointe, M.; Beaudet, A.; Howard, A.D. Expression of growth hormone secretagogue-receptors by growth hormone-releasing hormone neurons in the mediobasal hypothalamus. Endocrinology 1998, 139, 4420–4423.
- Hedegaard, M.A.; Holst, B. The Complex Signaling Pathways of the Ghrelin Receptor. Endocrinology 2020, 161.
- Ge, X.; Yang, H.; Bednarek, M.A.; Galon-Tilleman, H.; Chen, P.; Chen, M.; Lichtman, J.S.; Wang, Y.; Dalmas, O.; Yin, Y.; et al. LEAP2 Is an Endogenous Antagonist of the Ghrelin Receptor. Cell Metab. 2018, 27, 461–469.e6.
- Dixit, V.D.; Weeraratna, A.T.; Yang, H.; Bertak, D.; Cooper-Jenkins, A.; Riggins, G.J.; Eberhart, C.G.; Taub, D.D. Ghrelin and the growth hormone secretagogue receptor constitute a novel autocrine pathway in astrocytoma motility. J. Biol. Chem. 2006, 281, 16681–16690.
- Duxbury, M.S.; Waseem, T.; Ito, H.; Robinson, M.K.; Zinner, M.J.; Ashley, S.W.; Whang, E.E. Ghrelin promotes pancreatic adenocarcinoma cellular proliferation and invasiveness. Biochem. Biophys. Res. Commun. 2003, 309, 464–468.
- Chen, J.H.; Huang, S.M.; Chen, C.C.; Tsai, C.F.; Yeh, W.L.; Chou, S.J.; Hsieh, W.T.; Lu, D.Y. Ghrelin induces cell migration through GHS-R, CaMKII, AMPK, and NF-κB signaling pathway in glioma cells. J. Cell. Biochem. 2011, 112, 2931–2941.
- Hou, Z.; Miao, Y.; Gao, L.; Pan, H.; Zhu, S. Ghrelin-containing neuron in cerebral cortex and hypothalamus linked with the DVC of brainstem in rat. Regul. Pept. 2006, 134, 126–131.
- Raghay, K.; García-Caballero, T.; Nogueiras, R.; Morel, G.; Beiras, A.; Diéguez, C.; Gallego, R. Ghrelin localization in rat and human thyroid and parathyroid glands and tumours. Histochem. Cell Biol. 2006, 125, 239–246.
- Widmayer, P.; Partsch, V.; Pospiech, J.; Kusumakshi, S.; Boehm, U.; Breer, H. Distinct Cell Types With the Bitter Receptor Tas2r126 in Different Compartments of the Stomach. Front. Physiol. 2020, 11, 32.
- Date, Y.; Kojima, M.; Hosoda, H.; Sawaguchi, A.; Mondal, M.S.; Suganuma, T.; Matsukura, S.; Kangawa, K.; Nakazato, M. Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology 2000, 141, 4255–4261.
- Andralojc, K.M.; Mercalli, A.; Nowak, K.W.; Albarello, L.; Calcagno, R.; Luzi, L.; Bonifacio, E.; Doglioni, C.; Piemonti, L. Ghrelin-producing epsilon cells in the developing and adult human pancreas. Diabetologia 2009, 52, 486–493.
- Wierup, N.; Sundler, F.; Heller, R.S. The islet ghrelin cell. J. Mol. Endocrinol. 2013, 52, R35–R49.
- Sista, F.; Abruzzese, V.; Clementi, M.; Carandina, S.; Amicucci, G. Effect of Resected Gastric Volume on Ghrelin and GLP-1 Plasma Levels: A Prospective Study. J. Gastrointest. Surg. 2016, 20, 1931–1941.
- Dogan, U.; Ellidag, H.Y.; Aslaner, A.; Cakir, T.; Oruc, M.T.; Koc, U.; Mayir, B.; Gomceli, I.; Bulbuller, N.; Yilmaz, N. The impact of laparoscopic sleeve gastrectomy on plasma obestatin and ghrelin levels. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2113–2122.
- Camacho-Ramírez, A.; Mayo-Ossorio, M.Á.; Pacheco-García, J.M.; Almorza-Gomar, D.; Ribelles-García, A.; Belmonte-Núñez, A.; Prada-Oliveira, J.A.; Pérez-Arana, G.M. Pancreas is a preeminent source of ghrelin after sleeve gastrectomy in wistar rats. Histol. Histopathol. 2020, 35, 801–809.
- Takaya, K.; Ariyasu, H.; Kanamoto, N.; Iwakura, H.; Yoshimoto, A.; Harada, M.; Mori, K.; Komatsu, Y.; Usui, T.; Shimatsu, A.; et al. Ghrelin strongly stimulates growth hormone (GH) release in humans. J. Clin. Endocrinol. Metab. 2000, 85, 4908–4911.
- Broglio, F.; Benso, A.; Castiglioni, C.; Gottero, C.; Prodam, F.; Destefanis, S.; Gauna, C.; Van Der Lely, A.J.; Deghenghi, R.; Bo, M.; et al. The endocrine response to ghrelin as a function of gender in humans in young and elderly subjects. J. Clin. Endocrinol. Metab. 2003, 88, 1537–1542.
- Wren, A.M.; Seal, L.J.; Cohen, M.A.; Brynes, A.E.; Frost, G.S.; Murphy, K.G.; Dhillo, W.S.; Ghatei, M.A.; Bloom, S.R. Ghrelin enhances appetite and increases food intake in humans. J. Clin. Endocrinol. Metab. 2001, 86, 5992–5997.
- Wren, A.M.; Small, C.J.; Abbott, C.R.; Dhillo, W.S.; Seal, L.J.; Cohen, M.A.; Batterham, R.L.; Taheri, S.; Stanley, S.A.; Ghatei, M.A.; et al. Ghrelin causes hyperphagia and obesity in rats. Diabetes 2001, 50, 2540–2547.
- Kamegai, J.; Tamura, H.; Shimizu, T.; Ishii, S.; Sugihara, H.; Wakabayashi, I. Chronic Central Infusion of Ghrelin Increases Hypothalamic Neuropeptide Y and Agouti-Related Protein mRNA Levels and Body Weight in Rats. Diabetes 2001, 50, 2438–2443.
- Riediger, T.; Traebert, M.; Schmid, H.A.; Scheel, C.; Lutz, T.A.; Scharrer, E. Site-specific effects of ghrelin on the neuronal activity in the hypothalamic arcuate nucleus. Neurosci. Lett. 2003, 341, 151–155.
- Toshinai, K.; Date, Y.; Murakami, N.; Shimada, M.; Mondal, M.S.; Shimbara, T.; Guan, J.L.; Wang, Q.P.; Funahashi, H.; Sakurai, T.; et al. Ghrelin-induced food intake is mediated via the orexin pathway. Endocrinology 2003, 144, 1506–1512.
- Currie, P.J.; Coiro, C.D.; Duenas, R.; Guss, J.L.; Mirza, A.; Tal, N. Urocortin I inhibits the effects of ghrelin and neuropeptide Y on feeding and energy substrate utilization. Brain Res. 2011, 1385, 127–134.
- Stempniewicz, A.; Ceranowicz, P.; Warzecha, Z. Potential therapeutic effects of gut hormones, ghrelin and obestatin in oral mucositis. Int. J. Mol. Sci. 2019, 20, 1534.
- Foster-Schubert, K.E.; Overduin, J.; Prudom, C.E.; Liu, J.; Callahan, H.S.; Gaylinn, B.D.; Thorner, M.O.; Cummings, D.E. Acyl and total ghrelin are suppressed strongly by ingested proteins, weakly by lipids, and biphasically by carbohydrates. J. Clin. Endocrinol. Metab. 2008, 93, 1971–1979.
- Masuda, Y.; Tanaka, T.; Inomata, N.; Ohnuma, N.; Tanaka, S.; Itoh, Z.; Hosoda, H.; Kojima, M.; Kangawa, K. Ghrelin stimulates gastric acid secretion and motility in rats. Biochem. Biophys. Res. Commun. 2000, 276, 905–908.
- De La Cour, C.D.; Lindström, E.; Norlén, P.; Håkanson, R. Ghrelin stimulates gastric emptying but is without effect on acid secretion and gastric endocrine cells. Regul. Pept. 2004, 120, 23–32.
- Date, Y.; Murakami, N.; Toshinai, K.; Matsukura, S.; Niijima, A.; Matsuo, H.; Kangawa, K.; Nakazato, M. The role of the gastric afferent vagal nerve in Ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology 2002, 123, 1120–1128.
- Asakawa, A.; Inui, A.; Kaga, T.; Yuzuriha, H.; Nagata, T.; Ueno, N.; Makino, S.; Fujimiya, M.; Niijima, A.; Fujino, M.A.; et al. Ghrelin is an appetite-stimulatory signal from stomach with structural resemblance to motilin. Gastroenterology 2001, 120, 337–345.
- Yakabi, K.; Ro, S.; Onouhi, T.; Tanaka, T.; Ohno, S.; Miura, S.; Johno, Y.; Takayama, K. Histamine mediates the stimulatory action of ghrelin on acid secretion in rat stomach. Dig. Dis. Sci. 2006, 51, 1313–1321.
- Sakurada, T.; Ro, S.; Onouchi, T.; Ohno, S.; Aoyama, T.; Chinen, K.; Takabayashi, H.; Kato, S.; Takayama, K.; Yakabi, K. Comparison of the actions of acylated and desacylated ghrelin on acid secretion in the rat stomach. J. Gastroenterol. 2010, 45, 1111–1120.
- Fukumoto, K.; Nakahara, K.; Katayama, T.; Miyazatao, M.; Kangawa, K.; Murakami, N. Synergistic action of gastrin and ghrelin on gastric acid secretion in rats. Biochem. Biophys. Res. Commun. 2008, 374, 60–63.
- Levin, F.; Edholm, T.; Ehrström, M.; Wallin, B.; Schmidt, P.T.; Kirchgessner, A.M.; Hilsted, L.M.; Hellström, P.M.; Näslund, E. Effect of peripherally administered ghrelin on gastric emptying and acid secretion in the rat. Regul. Pept. 2005, 131, 59–65.
- Sibilia, V.; Pagani, F.; Guidobono, F.; Locatelli, V.; Torsello, A.; Deghenghi, R.; Netti, C. Evidence for a central inhibitory role of growth hormone secretagogues and ghrelin on gastric acid secretion in conscious rats. Neuroendocrinology 2002, 75, 92–97.
- Zhang, W.; Chen, M.; Chen, X.; Segura, B.J.; Mulholland, M.W. Inhibition of pancreatic protein secretion by ghrelin in the rat. J. Physiol. 2001, 537, 231–236.
- Sato, N.; Kanai, S.; Takano, S.; Kurosawa, M.; Funakoshi, A.; Miyasaka, K. Central Administration of Ghrelin Stimulates Pancreatic Exocrine Secretion via the Vagus in Conscious Rats. Jpn. J. Physiol. 2003, 53, 443–449.
- Lee, H.-M.; Wang, G.; Englander, E.W.; Kojima, M.; Greeley, G.H., Jr. Ghrelin, A New Gastrointestinal Endocrine Peptide that Stimulates Insulin Secretion: Enteric Distribution, Ontogeny, Influence of Endocrine, and Dietary Manipulations. Endocrinology 2002, 143, 185–190.
- Date, Y.; Nakazato, M.; Hashiguchi, S.; Dezaki, K.; Mondal, M.S.; Hosoda, H.; Kojima, M.; Kangawa, K.; Arima, T.; Matsuo, H.; et al. Ghrelin is present in pancreatic α-cells of humans and rats and stimulates insulin secretion. Diabetes 2002, 51, 124–129.
- Reimer, M.K.; Pacini, G.; Ahrén, B. Dose-dependent inhibition by ghrelin of insulin secretion in the mouse. Endocrinology 2003, 144, 916–921.
- Broglio, F.; Arvat, E.; Benso, A.; Gottero, C.; Muccioli, G.; Papotti, M.; van der Lely, A.J.; Deghenghi, R.; Ghigo, E. Ghrelin, a Natural GH Secretagogue Produced by the Stomach, Induces Hyperglycemia and Reduces Insulin Secretion in Humans. J. Clin. Endocrinol. Metab. 2001, 86, 5083.
- Dezaki, K. Ghrelin function in insulin release and glucose metabolism. Endocr. Dev. 2013, 25, 135–143.
- Lindqvist, A.; Shcherbina, L.; Prasad, R.B.; Miskelly, M.G.; Abels, M.; Martínez-Lopéz, J.A.; Fred, R.G.; Nergård, B.J.; Hedenbro, J.; Groop, L.; et al. Ghrelin suppresses insulin secretion in human islets and type 2 diabetes patients have diminished islet ghrelin cell number and lower plasma ghrelin levels. Mol. Cell. Endocrinol. 2020, 511, 110835.
- Dezaki, K.; Yada, T. Islet β-cell ghrelin signaling for inhibition of insulin secretion. Methods Enzymol. 2012, 514, 317–331.
- Kurashina, T.; Dezaki, K.; Yoshida, M.; Sukma Rita, R.; Ito, K.; Taguchi, M.; Miura, R.; Tominaga, M.; Ishibashi, S.; Kakei, M.; et al. The β-cell GHSR and downstream cAMP/TRPM2 signaling account for insulinostatic and glycemic effects of ghrelin. Sci. Rep. 2015, 5, 14041.
- Vestergaard, E.T.; Møller, N.; Andersen, R.F.; Rittig, S.; Jørgensen, J.O.L. Acute intravenous acyl ghrelin infusion induces thirst but does not affect sodium excretion: Two randomized, double-blind, placebo-controlled crossover studies in hypopituitary patients. Eur. J. Endocrinol. 2019, 181, 23–30.
- Han, Q.Q.; Huang, H.J.; Wang, Y.L.; Yang, L.; Pilot, A.; Zhu, X.C.; Yu, R.; Wang, J.; Chen, X.R.; Liu, Q.; et al. Ghrelin exhibited antidepressant and anxiolytic effect via the p38-MAPK signaling pathway in hippocampus. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 93, 11–20.
- Liu, G.B.; Pan, Y.M.; Liu, Y.S.; Hu, J.H.; Zhang, X.D.; Zhang, D.W.; Wang, Y.; Feng, Y.K.; Yu, J.B.; Cheng, Y.X. Ghrelin promotes neural differentiation of adipose tissue-derived mesenchymal stem cell via AKT/mTOR and β-catenin signaling pathways. Kaohsiung J. Med. Sci. 2020, 36, 405–416.
- Koutouratsas, T.; Kalli, T.; Karamanolis, G.; Gazouli, M. Contribution of ghrelin to functional gastrointestinal disorders’ pathogenesis. World J. Gastroenterol. 2019, 25, 539–551.
- Li, B.B.; Chen, Z.B.; Li, B.C.; Lin, Q.; Li, X.X.; Li, S.L.; Ding, C.; Wu, L.L.; Yu, G.Y. Expression of ghrelin in human salivary glands and its levels in saliva and serum in Chinese obese children and adolescents. Arch. Oral Biol. 2011, 56, 389–394.
- Aydin, S.; Ozercan, I.H.; Geckil, H.; Dagli, F.; Aydin, S.; Kumru, S.; Kilic, N.; Sahin, I.; Ozercan, M.R. Ghrelin is present in teeth. J. Biochem. Mol. Biol. 2007, 40, 368–372.
- Ohta, K.; Laborde, N.J.; Kajiya, M.; Shin, J.; Zhu, T.; Thondukolam, A.K.; Min, C.; Kamata, N.; Karimbux, N.Y.; Stashenko, P.; et al. Expression and possible immune-regulatory function of ghrelin in oral epithelium. J. Dent. Res. 2011, 90, 1286–1292.
- Shin, Y.K.; Martin, B.; Kim, W.; White, C.M.; Ji, S.; Sun, Y.; Smith, R.G.; Sévigny, J.; Tschöp, M.H.; Maudsley, S.; et al. Ghrelin is produced in taste cells and ghrelin receptor null mice show reduced taste responsivity to salty (NaCl) and sour (citric acid) tastants. PLoS ONE 2010, 5, e0121327.
- Liu, B.; Han, X.; Feng, W.; Cui, J.; Hasegawa, T.; Amizuka, N.; Xu, X.; Li, M. Altered distribution of Ghrelin protein in mice molar development. Arch. Oral Biol. 2016, 65, 82–86.
- Gröschl, M.; Topf, H.G.; Bohlender, J.; Zenk, J.; Klussmann, S.; Dötsch, J.; Rascher, W.; Rauh, M. Identification of ghrelin in human saliva: Production by the salivary glands and potential role in proliferation of oral keratinocytes. Clin. Chem. 2005, 51, 997–1006.
- Aydin, S.; Halifeoglu, I.; Ozercan, I.H.; Erman, F.; Kilic, N.; Aydin, S.; Ilhan, N.; Ilhan, N.; Ozkan, Y.; Akpolat, N.; et al. A comparison of leptin and ghrelin levels in plasma and saliva of young healthy subjects. Peptides 2005, 26, 647–652.
- Nokhbehsaim, M.; Memmert, S.; Damanaki, A.; Nanayakkara, S.; Zhou, X.; Jäger, A.; Deschner, J. Effect of interleukin-1β on ghrelin receptor in periodontal cells. Clin. Oral Investig. 2019, 23, 113–122.
- Warzecha, Z.; Kownacki, P.; Ceranowicz, P.; Dembinski, M.; Cieszkowski, J.; Dembinski, A. Ghrelin accelerates the healing of oral ulcers in non-sialoadenectomized and sialoadenectomized rats. J. Physiol. Pharmacol. 2013, 64, 657–668.
- Konturek, P.C.; Burnat, G.; Rau, T.; Hahn, E.G.; Konturek, S. Effect of adiponectin and ghrelin on apoptosis of Barrett adenocarcinoma cell line. Dig. Dis. Sci. 2008, 53, 597–605.
- Thomas, S.J.; Almers, L.; Schneider, J.; Graham, J.E.; Havel, P.J.; Corley, D.A. Ghrelin and Leptin Have a Complex Relationship with Risk of Barrett’s Esophagus. Dig. Dis. Sci. 2016, 61, 70–79.
- Takata, A.; Takiguchi, S.; Miyazaki, Y.; Miyata, H.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; Nakajima, K.; Mori, M.; Kangawa, K.; et al. Randomized Phase II Study of the Anti-inflammatory Effect of Ghrelin During the Postoperative Period of Esophagectomy. Ann. Surg. 2015, 262, 230–236.
- Yamashita, K.; Yamamoto, K.; Takata, A.; Miyazaki, Y.; Saito, T.; Tanaka, K.; Makino, T.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; et al. Continuous ghrelin infusion attenuates the postoperative inflammatory response in patients with esophageal cancer. Esophagus 2021, 18, 239–247.
- Yamamoto, K.; Takiguchi, S.; Miyata, H.; Miyazaki, Y.; Hiura, Y.; Yamasaki, M.; Nakajima, K.; Fujiwara, Y.; Mori, M.; Kangawa, K.; et al. Reduced plasma ghrelin levels on day 1 after esophagectomy: A new predictor of prolonged systemic inflammatory response syndrome. Surg. Today 2013, 43, 48–54.
- Murphy, G.; Kamangar, F.; Dawsey, S.M.; Stanczyk, F.Z.; Weinstein, S.J.; Taylor, P.R.; Virtamo, J.; Abnet, C.C.; Albanes, D.; Freedman, N.D. The relationship between serum ghrelin and the risk of gastric and esophagogastric junctional adenocarcinomas. J. Natl. Cancer Inst. 2011, 103, 1123–1129.
- Sibilia, V.; Rindi, G.; Pagani, F.; Rapetti, D.; Locatelli, V.; Torsello, A.; Campanini, N.; Deghenghi, R.; Netti, C. Ghrelin protects against ethanol-induced gastric ulcers in rats: Studies on the mechanisms of action. Endocrinology 2003, 144, 353–359.
- Konturek, P.C.; Brzozowski, T.; Pajdo, R.; Nikiforuk, A.; Kwiecien, S.; Harsch, I.; Drozdowicz, D.; Hahn, E.G.; Konturek, S.J. Ghrelin—A new gastroprotective factor in gastric mucosa. J. Physiol. Pharmacol. 2004, 55, 325–336.
- Brzozowski, T.; Konturek, P.C.; Konturek, S.J.; Kwiecień, S.; Drozdowicz, D.; Bielanski, W.; Pajdo, R.; Ptak, A.; Nikiforuk, A.; Pawlik, W.W.; et al. Exogenous and endogenous ghrelin in gastroprotection against stress-induced gastric damage. Regul. Pept. 2004, 120, 39–51.
- Brzozowski, T.; Konturek, P.C.; Drozdowicz, D.; Konturek, S.J.; Pawlik, M.; Sliwowski, Z.; Pawlik, W.W.; Hahn, E.G. Role of central and peripheral ghrelin in the mechanism of gastric mucosal defence. Inflammopharmacology 2005, 13, 45–62.
- Konturek, P.C.; Brzozowski, T.; Walter, B.; Burnat, G.; Hess, T.; Hahn, E.G.; Konturek, S.J. Ghrelin-induced gastroprotection against ischemia-reperfusion injury involves an activation of sensory afferent nerves and hyperemia mediated by nitric oxide. Eur. J. Pharmacol. 2006, 536, 171–181.
- Brzozowski, T.; Konturek, P.C.; Sliwowski, Z.; Pajdo, R.; Drozdowicz, D.; Kwiecien, S.; Burnat, G.; Konturek, S.J.; Pawlik, W.W. Prostaglandin/cyclooxygenase pathway in ghrelin-induced gastroprotection against ischemia-reperfusion injury. J. Pharmacol. Exp. Ther. 2006, 319, 477–487.
- Adami, M.; Pozzoli, C.; Leurs, R.; Stark, H.; Coruzzi, G. Histamine H(3) receptors are involved in the protective effect of ghrelin against HCl-induced gastric damage in rats. Pharmacology 2010, 86, 259–266.
- Íşeri, S.Ö.; Şener, G.; Yüksel, M.; Contuk, G.; Çetinel, Ş.; Gedik, N.; Yeǧen, B.Ç. Ghrelin against alendronate-induced gastric damage in rats. J. Endocrinol. 2005, 187, 399–406.
- Warzecha, Z.; Ceranowicz, P.; Dembinski, M.; Cieszkowski, J.; Ginter, G.; Ptak-Belowska, A.; Dembinski, A. Involvement of cyclooxygenase-1 and cyclooxygenase-2 activity in the therapeutic effect of ghrelin in the course of ethanol-induced gastric ulcers in rats. J. Physiol. Pharmacol. 2014, 65, 95–106.
- Ceranowicz, P.; Warzecha, Z.; Dembinski, A.; Sendur, R.; Cieszkowski, J.; Ceranowicz, D.; Pawlik, W.W.; Kuwahara, A.; Kato, I.; Konturek, P.C. Treatment with ghrelin accelerates the healing of acetic acid-induced gastric and duodenal ulcers in rats. J. Physiol. Pharmacol. 2009, 60, 87–98.
- Wu, R.; Dong, W.; Ji, Y.; Zhou, M.; Marini, C.P.; Ravikumar, T.S.; Wang, P. Orexigenic hormone ghrelin attenuates local and remote organ injury after intestinal ischemia-reperfusion. PLoS ONE 2008, 3, e2026.
- Pawlik, M.W.; Obuchowicz, R.; Biernat, J.; Szczepanski, W.; Pajdo, R.; Kwiecień, S.; Brzozowski, T.; Konturek, S.J.; Pawlik, W.W. Effects of peripherally and centrally applied ghrelin in the pathogenesis of ischemia-reperfusion induced injury of the small intestine. J. Physiol. Pharmacol. 2011, 62, 429–439.
- Zhang, H.; Cui, Z.; Luo, G.; Zhang, J.; Ma, T.; Hu, N.; Cui, T. Ghrelin attenuates intestinal ischemia/reperfusion injury in mice by activating the mTOR signaling pathway. Int. J. Mol. Med. 2013, 32, 851–859.
- Wang, Z.; Yang, W.L.; Jacob, A.; Aziz, M.; Wang, P. Human ghrelin mitigates intestinal injury and mortality after whole body irradiation in rats. PLoS ONE 2015, 10, e0118213.
- Cheng, Y.; Wei, Y.; Yang, W.; Cai, Y.; Chen, B.; Yang, G.; Shang, H.; Zhao, W. Ghrelin Attenuates Intestinal Barrier Dysfunction Following Intracerebral Hemorrhage in Mice. Int. J. Mol. Sci. 2016, 17, 2032.
- Warzecha, Z.; Ceranowicz, D.; Dembiński, A.; Ceranowicz, P.; Cieszkowski, J.; Kuwahara, A.; Kato, I.; Dembiński, M.; Konturek, P.C. Ghrelin accelerates the healing of cysteamine-induced duodenal ulcers in rats. Med. Sci. Monit. 2012, 18, BR181.
- Onishi, S.; Kaji, T.; Yamada, W.; Nakame, K.; Machigashira, S.; Kawano, M.; Yano, K.; Harumatsu, T.; Yamada, K.; Masuya, R.; et al. Ghrelin stimulates intestinal adaptation following massive small bowel resection in parenterally fed rats. Peptides 2018, 106, 59–67.
- Mendez-Sanchez, N.; Ponciano-Rodriguez, G.; Bermejo-Martinez, L.; Villa, A.R.; Chavez-Tapia, N.C.; Zamora-Valdes, D.; Pichardo-Bahena, R.; Barredo-Prieto, B.; Uribe-Ramos, M.H.; Ramos, M.H.; et al. Low serum levels of ghrelin are associated with gallstone disease. World J. Gastroenterol. 2006, 12, 3096–3100.
- Gutierrez-Grobe, Y.; Villalobos-Blasquez, I.; Sánchez-Lara, K.; Villa, A.R.; Ponciano-Rodríguez, G.; Ramos, M.H.; Chavez-Tapia, N.C.; Uribe, M.; Méndez-Sánchez, N. High ghrelin and obestatin levels and low risk of developing fatty liver. Ann. Hepatol. 2010, 9, 52–57.
- Ezquerro, S.; Mocha, F.; Frühbeck, G.; Guzmán-Ruiz, R.; Valentí, V.; Mugueta, C.; Becerril, S.; Catalán, V.; Gómez-Ambrosi, J.; Silva, C.; et al. Ghrelin Reduces TNF-α-Induced Human Hepatocyte Apoptosis, Autophagy, and Pyroptosis: Role in Obesity-Associated NAFLD. J. Clin. Endocrinol. Metab. 2019, 104, 21–37.
- Nagoya, T.; Kamimura, K.; Inoue, R.; Ko, M.; Owaki, T.; Niwa, Y.; Sakai, N.; Setsu, T.; Sakamaki, A.; Yokoo, T.; et al. Ghrelin-insulin-like growth factor-1 axis is activated via autonomic neural circuits in the non-alcoholic fatty liver disease. Neurogastroenterol. Motil. 2020, 32.
- Li, Y.; Hai, J.; Li, L.; Chen, X.; Peng, H.; Cao, M.; Zhang, Q. Administration of ghrelin improves inflammation, oxidative stress, and apoptosis during and after non-alcoholic fatty liver disease development. Endocrine 2013, 43, 376–386.
- Golestan Jahromi, M.; Nabavizadeh, F.; Vahedian, J.; Nahrevanian, H.; Dehpour, A.R.; Zare-Mehrjardi, A. Protective effect of ghrelin on acetaminophen-induced liver injury in rat. Peptides 2010, 31, 2114–2117.
- Moreno, M.; Chaves, J.F.; Sancho-Bru, P.; Ramalho, F.; Ramalho, L.N.; Mansego, M.L.; Ivorra, C.; Dominguez, M.; Conde, L.; Millán, C.; et al. Ghrelin attenuates hepatocellular injury and liver fibrogenesis in rodents and influences fibrosis progression in humans. Hepatology 2010, 51, 974–985.
- Qin, Y.; Li, Z.; Wang, Z.; Li, Y.; Zhao, J.; Mulholland, M.; Zhang, W. Ghrelin contributes to protection of hepatocellular injury induced by ischaemia/reperfusion. Liver Int. 2014, 34, 567–575.
- Cetin, E.; Kanbur, M.; Cetin, N.; Eraslan, G.; Atasever, A. Hepatoprotective effect of ghrelin on carbon tetrachloride-induced acute liver injury in rats. Regul. Pept. 2011, 171, 1–5.
- Arıcı, O.F.; Cetin, N. Protective role of ghrelin against carbon tetrachloride (CCl₄)-induced coagulation disturbances in rats. Regul. Pept. 2011, 166, 139–142.
- Granata, R.; Settanni, F.; Trovato, L.; Destefanis, S.; Gallo, D.; Martinetti, M.; Ghigo, E.; Muccioli, G. Unacylated as well as acylated ghrelin promotes cell survival and inhibit apoptosis in HIT-T15 pancreatic beta-cells. J. Endocrinol. Investig. 2006, 29.
- Granata, R.; Settanni, F.; Biancone, L.; Trovato, L.; Nano, R.; Bertuzzi, F.; Destefanis, S.; Annunziata, M.; Martinetti, M.; Catapano, F.; et al. Acylated and unacylated ghrelin promote proliferation and inhibit apoptosis of pancreatic beta-cells and human islets: Involvement of 3’,5’-cyclic adenosine monophosphate/protein kinase A, extracellular signal-regulated kinase 1/2, and phosphatidyl inositol 3-Kinase/Akt signaling. Endocrinology 2007, 148, 512–529.
- Granata, R.; Volante, M.; Settanni, F.; Gauna, C.; Ghé, C.; Annunziata, M.; Deidda, B.; Gesmundo, I.; Abribat, T.; van der Lely, A.J.; et al. Unacylated ghrelin and obestatin increase islet cell mass and prevent diabetes in streptozotocin-treated newborn rats. J. Mol. Endocrinol. 2010, 45, 9–17.
- Wang, W.; Zhang, D.; Zhao, H.; Chen, Y.; Liu, Y.; Cao, C.; Han, L.; Liu, G. Ghrelin inhibits cell apoptosis induced by lipotoxicity in pancreatic beta-cell line. Regul. Pept. 2010, 161, 43–50.
- Diaz-Ganete, A.; Baena-Nieto, G.; Lomas-Romero, I.M.; Lopez-Acosta, J.F.; Cozar-Castellano, I.; Medina, F.; Segundo, C.; Lechuga-Sancho, A.M. Ghrelin’s Effects on Proinflammatory Cytokine Mediated Apoptosis and Their Impact on β-Cell Functionality. Int. J. Endocrinol. 2015, 2015.
- Baena-Nieto, G.; Lomas-Romero, I.M.; Mateos, R.M.; Leal-Cosme, N.; Perez-Arana, G.; Aguilar-Diosdado, M.; Segundo, C.; Lechuga-Sancho, A.M. Ghrelin mitigates β-cell mass loss during insulitis in an animal model of autoimmune diabetes mellitus, the BioBreeding/Worcester rat. Diabetes Metab. Res. Rev. 2017, 33.
- Dembinski, A.; Warzecha, Z.; Ceranowicz, P.; Tomaszewska, R.; Stachura, J.; Konturek, S.J.; Konturek, P.C. Ghrelin attenuates the development of acute pancreatitis in rats. J. Physiol. Pharmacol. 2003, 54, 561–573.
- Dembiński, A.; Warzecha, Z.; Ceranowicz, P.; Cieszkowski, J.; Pawlik, W.W.; Tomaszewska, R.; Kuśnierz-Cabala, B.; Naskalski, J.W.; Kuwahara, A.; Kato, I. Role of growth hormone and insulin-like growth factor-1 in the protective effect of ghrelin in ischemia/reperfusion-induced acute pancreatitis. Growth Horm. IGF Res. 2006, 16, 348–356.
- Zhou, X.; Xue, C. Ghrelin attenuates acute pancreatitis-induced lung injury and inhibits substance P expression. Am. J. Med. Sci. 2010, 339, 49–54.
- Zhou, X.; Xue, C. Ghrelin inhibits the development of acute pancreatitis and nuclear factor kappaB activation in pancreas and liver. Pancreas 2009, 38, 752–757.
- Bonior, J.; Ceranowicz, P.; Gajdosz, R.; Kuśnierz-Cabala, B.; Pierzchalski, P.; Warzecha, Z.; Dembiński, A.; Pędziwiatr, M.; Kot, M.; Szpak, A.L.; et al. Molecular ghrelin system in the pancreatic acinar cells: The role of the polypeptide, caerulein and sensory nerves. Int. J. Mol. Sci. 2017, 18, 929.
- Chang, R.J.; Wang, H.L.; Qin, M.B.; Liang, Z.H.; He, J.P.; Wei, Y.L.; Fu, H.Z.; Tang, G.D. Ghrelin inhibits IKKβ/NF-κB activation and reduces pro-inflammatory cytokine production in pancreatic acinar AR42J cells treated with cerulein. Hepatobiliary Pancreat. Dis. Int. 2020, 20, 366–375.
- Bonior, J.; Warzecha, Z.; Ceranowicz, P.; Gajdosz, R.; Pierzchalski, P.; Kot, M.; Leja-Szpak, A.; Nawrot-Porąbka, K.; Link-Lenczowski, P.; Pędziwiatr, M.; et al. Capsaicin-Sensitive Sensory Nerves Are Necessary for the Protective Effect of Ghrelin in Cerulein-Induced Acute Pancreatitis in Rats. Int. J. Mol. Sci. 2017, 18, 1402.
- Tang, X.; Tang, G.; Liang, Z.; Qin, M.; Fang, C.; Zhang, L. Effects of Ghrelin miRNA on Inflammation and Calcium Pathway in Pancreatic Acinar Cells of Acute Pancreatitis. Pancreas 2017, 46, 1305–1313.
- Warzecha, Z.; Ceranowicz, P.; Dembinski, A.; Cieszkowski, J.; Kusnierz-Cabala, B.; Tomaszewska, R.; Kuwahara, A.; Kato, I. Therapeutic effect of ghrelin in the course of cerulein induced acute pancreatitis in rats. J. Physiol. Pharmacol. 2010, 61, 419–427.
- Bukowczan, J.; Warzecha, Z.; Ceranowicz, P.; Kusnierz-Cabala, B.; Tomaszewska, R.; Dembinski, A. Therapeutic effect of ghrelin in the course of ischemia/reperfusion-induced acute pancreatitis. Curr. Pharm. Des. 2015, 21, 2284–2290.
- Wang, H.; Qin, M.; Liang, Z.; Chang, R.; Fu, H.; Wei, Y.; Tang, G. Serum ghrelin, but not obestatin, is a potential predictor of acute pancreatitis severity. Medicine 2017, 96, e7963.
- Panek, J.; Bonior, J.; Pieton, J.; Jaworek, J. Serum leptin and ghrelin levels in patients in the early stages of acute biliary pancreatitis and different degrees of severity. Pol. Przegl. Chir. 2014, 86, 211–217.
- Daniel, P.; Leśniowski, B.; Jasińska, A.; Pietruczuk, M.; Małecka-Panas, E. Usefulness of assessing circulating levels of resistin, ghrelin, and IL-18 in alcoholic acute pancreatitis. Dig. Dis. Sci. 2010, 55, 2982–2987.
- Türkoğlu, A.; Böyük, A.; Tanrıverdi, M.H.; Gündüz, E.; Dusak, A.; Kaplan, İ.; Gümüş, M. The potential role of BMI, plasma leptin, nesfatin-1 and ghrelin levels in the early detection of pancreatic necrosis and severe acute pancreatitis: A prospective cohort study. Int. J. Surg. 2014, 12, 1310–1313.
- Sasaki, K.; Asaoka, T.; Eguchi, H.; Fukuda, Y.; Iwagami, Y.; Yamada, D.; Miyazaki, Y.; Noda, T.; Takahashi, T.; Gotoh, K.; et al. Plasma ghrelin suppression as an early predictor for postoperative complications after pancreatoduodenectomy. Pancreatology 2018, 18, 73–78.
- Kerem, M.; Salman, B.; Ozsoy, S.; Pasaoglu, H.; Bedirli, A.; Haziroglu, R.; Yilmaz, T.U. Exogenous ghrelin enhances endocrine and exocrine regeneration in pancreatectomized rats. J. Gastrointest. Surg. 2009, 13, 775–783.
- Ates, Y.; Degertekin, B.; Erdil, A.; Yaman, H.; Dagalp, K. Serum ghrelin levels in inflammatory bowel disease with relation to disease activity and nutritional status. Dig. Dis. Sci. 2008, 53, 2215–2221.
- Karmiris, K.; Koutroubakis, I.E.; Xidakis, C.; Polychronaki, M.; Voudouri, T.; Kouroumalis, E.A. Circulating levels of leptin, adiponectin, resistin, and ghrelin in inflammatory bowel disease. Inflamm. Bowel Dis. 2006, 12, 100–105.
- Peracchi, M.; Bardella, M.T.; Caprioli, F.; Massironi, S.; Conte, D.; Valenti, L.; Ronchi, C.; Beck-Peccoz, P.; Arosio, M.; Piodi, L. Circulating ghrelin levels in patients with inflammatory bowel disease. Gut 2006, 55, 432–433.
- Hosomi, S.; Oshitani, N.; Kamata, N.; Sogawa, M.; Yamagami, H.; Watanabe, K.; Tominaga, K.; Watanabe, T.; Fujiwara, Y.; Maeda, K.; et al. Phenotypical and functional study of ghrelin and its receptor in the pathogenesis of Crohn’s disease. Inflamm. Bowel Dis. 2008, 14, 1205–1213.
- Konturek, P.C.; Brzozowski, T.; Engel, M.; Burnat, G.; Gaca, P.; Kwiecien, S.; Pajdo, R.; Konturek, S.J. Ghrelin ameliorates colonic inflammation. Role of nitric oxide and sensory nerves. J. Physiol. Pharmacol. 2009, 60, 41–47.
- Gonzalez-Rey, E.; Chorny, A.; Delgado, M. Therapeutic Action of Ghrelin in a Mouse Model of Colitis. Gastroenterology 2006, 130, 1707–1720.
- Zhang, L.; Cheng, J.; Shen, J.; Wang, S.; Guo, C.; Fan, X. Ghrelin Inhibits Intestinal Epithelial Cell Apoptosis Through the Unfolded Protein Response Pathway in Ulcerative Colitis. Front. Pharmacol. 2021, 12, 405.
- Maduzia, D.; Matuszyk, A.; Ceranowicz, D.; Warzecha, Z.; Ceranowicz, P.; Fyderek, K.; Galazka, K.; Dembinski, A. The influence of pretreatment with ghrelin on the development of acetic-acid-induced colitis in rats. J. Physiol. Pharmacol. 2015, 66, 875–885.
- Matuszyk, A.; Ceranowicz, P.; Warzecha, Z.; Cieszkowski, J.; Ceranowicz, D.; Gałązka, K.; Bonior, J.; Jaworek, J.; Bartuś, K.; Gil, K.; et al. Exogenous ghrelin accelerates the healing of acetic acid-induced colitis in rats. Int. J. Mol. Sci. 2016, 17, 1455.
- Ceranowicz, P.; Warzecha, Z.; Cieszkowski, J.; Ceranowicz, D.; Kúsnierz-Cabala, B.; Bonior, J.; Jaworek, J.; Ambroży, T.; Gil, K.; Olszanecki, R.; et al. Essential role of growth hormone and IGF-1 in therapeutic effect of ghrelin in the course of acetic acid-induced colitis. Int. J. Mol. Sci. 2017, 18, 1118.
- Ozturk, C.C.; Oktay, S.; Yuksel, M.; Akakin, D.; Yarat, A.; Kasimay Cakir, O. Anti-inflammatory effects of nesfatin-1 in rats with acetic acid-induced colitis and underlying mechanisms. J. Physiol. Pharmacol. 2015, 66, 741–750.
- Matuszyk, A.; Ceranowicz, D.; Warzecha, Z.; Ceranowicz, P.; Fyderek, K.; Gałązka, K.; Cieszkowski, J.; Bonior, J.; Jaworek, J.; Pihut, M.; et al. The Influence of Ghrelin on the Development of Dextran Sodium Sulfate-Induced Colitis in Rats. BioMed Res. Int. 2015, 2015, 718314.
- Cheng, J.; Zhang, L.; Dai, W.; Mao, Y.; Li, S.; Wang, J.; Li, H.; Guo, C.; Fan, X. Ghrelin ameliorates intestinal barrier dysfunction in experimental colitis by inhibiting the activation of nuclear factor-kappa B. Biochem. Biophys. Res. Commun. 2015, 458, 140–147.
- De Smet, B.; Thijs, T.; Moechars, D.; Colsoul, B.; Polders, L.; Ver Donck, L.; Coulie, B.; Peeters, T.L.; Depoortere, I. Endogenous and exogenous ghrelin enhance the colonic and gastric manifestations of dextran sodium sulphate-induced colitis in mice. Neurogastroenterol. Motil. 2009, 21, 59–70.
- Liu, Z.Z.; Wang, W.G.; Li, Q.; Tang, M.; Li, J.; Wu, W.T.; Wan, Y.H.; Wang, Z.G.; Bao, S.S.; Fei, J. Growth hormone secretagogue receptor is important in the development of experimental colitis. Cell Biosci. 2015, 5, 1–10.
- Tian, P.; Lu, X.; Jin, N.; Shi, J. Knockdown of ghrelin-O-acyltransferase attenuates colitis through the modulation of inflammatory factors and tight junction proteins in the intestinal epithelium. Cell Biol. Int. 2020, 44, 1681–1690.