Neuroblastoma is one of the most common childhood solid tumors and develops from neural stem cells that normally comprise the embryonic structure termed the neural crest. Human neuroblastoma cell lines have special properties as they exhibit cell growth and are induced to become mature neurons by drugs such as retinoid.
- neuroblastoma
- asymmetric cell division
- centrosome
- MYCN
- TRIM32
- cancer
1. Introduction
Neuroblastoma is one of the most typical solid tumors among childhood cancers and exhibits a wide clinical range [1][2][3]. Patients can be broadly divided into three patterns (low-risk, medium-risk, and high-risk groups) from a biological and clinical point of view [1][2][3]. Although minimum treatment may be sufficient for low-risk patients, high risk patents have poor results in spite of aggressive treatment. Among several biological features, amplification of the MYCN gene is known to correlate with its prognosis and malignancy. In fact, approximately 20% of neuroblastomas have MYCN gene amplification. Recent studies showed that MYCN not only exhibits oncogenic activity, but also plays a role in the self-renewal of normal neural stem cells and progenitor cells [4][5][6][7].
Neuroblastoma is believed to be derived from the multipotent neural crest cells that make up the structure of the embryo [8]. The neural crest is composed of a cell population that produce cell lineage such as Schwann cell, melanocyte, smooth muscle, peripheral neurons, and glia [8]. Therefore, neuroblastoma cells are suspected to be similar in nature to cancer stem cells due to their pluripotent function.
Cancer stem cells are thought to exhibit asymmetric cell division (ACD), which results in tumor cell heterogeneity [9][10]. ACD is a originate physiological process to maintain the stem cell pool and differentiated cell pool through a single cell division. Recent studies showed that the imbalance between self-renewal and differentiation causes the appearance of abnormal stem cells, leading to tumorigenesis in the Drosophila neuroblast population [11][12][13]. Thus, ACD is a strategy for producing many cancer stem cells and differentiated cancer cells.
2. Discovery of ACD in Human Neuroblastoma Cells
ACD studies were originally demonstrated using model organisms, such as nematode embryos [14][15], Drosophila neuroblasts [16], and Drosophila male germ stem cells [17]. These genetic studies showed that the mechanism of ACD is highly conserved. Thereafter, evidence supporting asymmetric cell division in mammalian stem cells, such as muscle [18], skin [19], gut [20], mammary glands [21], the hematopoietic system [22], and developing mouse brain [23][24], was reported.
As mentioned above, human neuroblastoma cell lines have special properties as they exhibit cell growth and are induced to become mature neurons by drugs such as retinoid [25]. In addition, neuroblastoma is a common childhood cancer that may develop at the fetal development stage when a large number of stem cells display ACD [26]. Therefore, we investigate ACD using human neuroblastoma cells as a model [27]. As a result, although the cell lines with MYCN amplification exhibited symmetric cell division (self-renewal division), the cell lines without MYCN amplification exhibited ACD ( Figure 1 ). In addition, comparison of ACD studies using these organisms and model systems demonstrated that ACD in neuroblastoma cells is evolutionarily conserved [27].
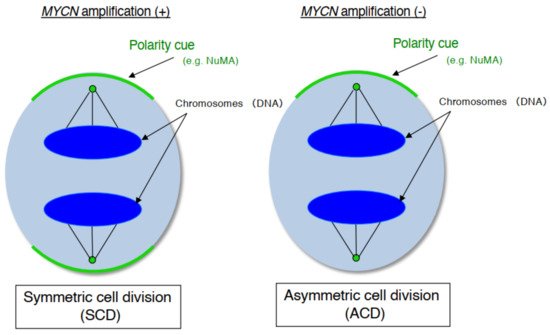
During late mitosis (anaphase), spindle pole-localized NuMA also localizes to the cell cortex as a polarity cue, and the cell cortex localized-NuMA ensures proper polarity division. Neuroblastoma cells with MYCN amplification exhibit symmetric NuMA cortex-based cell division, whereas those without MYCN amplification exhibit asymmetric NuMA cortex-based cell division (the ACD ratio is approximately 20%).
3. MYCN Regulates Cell Division Fate
MYCN is a typical transcriptional factor that belongs to the MYC family (c-MYC, L-MYC) [28]. As mentioned above, recent studies have reported that MYCN not only exhibits oncogenic activity, but also plays a central role in the self-renewal of normal neural stem and progenitor cells [29].
In our experiments, the gene expression level of MYCN influenced the control of cell division fate. The overexpression of MYCN induced symmetric cell division (SCD) (self-renewal division), whereas decreased expression of MYCN caused ACD [27]. Moreover, when its leucine zipper domain was deleted, the MYCN mutant no longer induced self-renewal division. This suggested that the transcriptional activity of MYCN is necessary for inducing SCD in human neuroblastoma cells [27]. Although the specific transcriptional target(s) of MYCN currently remain unclear except for the high mobility group A1 (HMGA1) oncogene [30], several molecular pathways have been identified in MYCN-mediated cell division fate ( Figure 2 ) [26].
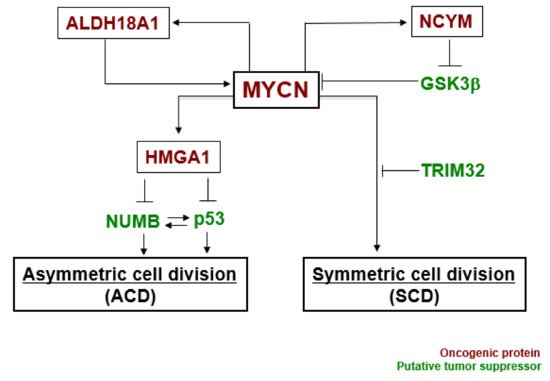
MYCN and ALDH18A1 form a positive feedback loop for their transcriptional expression [31]. This positive feedback loop effectively induces symmetric cell division (SCD). NCYM inhibits GSK3β --mediated phosphorylation of MYCN, resulting in induction of SCD [32][33]. HMGA1 is a transcriptional target of MYCN [30]. HMGA1 inhibits the expression of NUMB [34] and p53 [35]. NUMB and p53 induce asymmetric cell division (ACD) [21][36]. Lastly, TRIM32 facilitates proteasomal degradation of MYCN to induce asymmetric cell division (ACD) [37]. Thus, MYCN-dependent tumor cells exhibit symmetric cell division (SCD) and the degradation of MYCN causes asymmetric cell division (ACD).
4. Induction of ACD in Human Neuroblastoma Cells
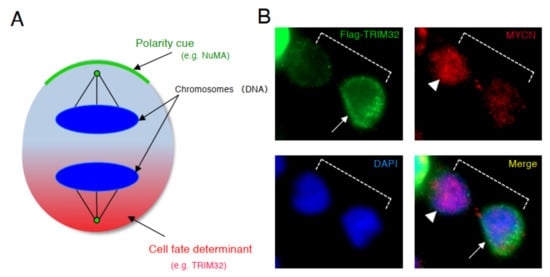
Based on the results of above previous studies, we searched for a molecule that degrades the MYCN protein and induces asymmetric division, focusing on TRIM32 (Tripartite motif-containing 32) by analogy. Previous studies reported that TRIM32 , an ortholog of Drosophila melanogaster , Brat , which controls ACD as a determinant of neuron, suppresses Drosophila MYC (dMYC) function in the neuroblasts of flies [38]. Moreover, mouse TRIM32 was reported to have ubiquitin ligase activity, and facilitated the degradation of the c-MYC oncoprotein during neurogenesis [39]. In neuroblastoma cells, TRIM32 associated with MYCN at the spindle pole during mitosis and facilitated the degradation of MYCN protein as an ubiquitin ligase [37]. In addition, when TRIM32 was overexpressed, ACD was detected in human neuroblastoma cells, as expected ( Figure 3 ) [37].
This entry is adapted from the peer-reviewed paper 10.3390/sym13101907
References
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216.
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078.
- Nakagawara, A.; Li, Y.; Izumi, H.; Muramori, K.; Inada, H.; Nishi, M. Neuroblastoma. Jpn. J. Clin. Oncol. 2018, 48, 214–241.
- Cotterman, R.; Knoepfler, P.S. N-Myc regulates expression of pluripotency genes in neuroblastoma including lif, klf2, klf4, and lin28b. PLoS ONE 2009, 4, e5799.
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415.
- Zhang, J.T.; Weng, Z.H.; Tsang, K.S.; Tsang, L.L.; Chan, H.C.; Jiang, X.H. MycN Is Critical for the Maintenance of Human Embryonic Stem Cell-Derived Neural Crest Stem Cells. PLoS ONE 2016, 11, e0148062.
- Kerosuo, L.; Neppala, P.; Hsin, J.; Mohlin, S.; Vieceli, F.M.; Torok, Z.; Laine, A.; Westermarck, J.; Bronner, M.E. Enhanced expression of MycN/CIP2A drives neural crest toward a neural stem cell-like fate: Implications for priming of neuroblastoma. Proc. Natl. Acad. Sci. USA 2018, 115, E7351–E7360.
- Ross, R.A.; Spengler, B.A. Human neuroblastoma stem cells. Semin. Cancer Biol. 2007, 17, 241–247.
- Morrison, S.J.; Kimble, J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 2006, 441, 1068–1074.
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337.
- Caussinus, E.; Gonzalez, C. Induction of tumor growth by altered stem-cell asymmetric division in Drosophila melanogaster. Nat. Genet. 2005, 37, 1125–1129.
- Clevers, H. Stem cells, asymmetric division and cancer. Nat. Genet. 2005, 37, 1027–1028.
- Knoblich, J.A. Asymmetric cell division: Recent developments and their implications for tumour biology. Nat. Rev. Mol. Cell Biol. 2010, 11, 849–860.
- Gonczy, P. Mechanisms of asymmetric cell division: Flies and worms pave the way. Nat. Rev. Mol. Cell Biol. 2008, 9, 355–366.
- Sawa, H. Control of cell polarity and asymmetric division in C. elegans. Curr. Top. Dev. Biol. 2012, 101, 55–76.
- Gonzalez, C. Spindle orientation, asymmetric division and tumour suppression in Drosophila stem cells. Nat. Rev. Genet. 2007, 8, 462–472.
- Yamashita, Y.M.; Mahowald, A.P.; Perlin, J.R.; Fuller, M.T. Asymmetric inheritance of mother versus daughter centrosome in stem cell division. Science 2007, 315, 518–521.
- Shinin, V.; Gayraud-Morel, B.; Gomes, D.; Tajbakhsh, S. Asymmetric division and cosegregation of template DNA strands in adult muscle satellite cells. Nat. Cell Biol. 2006, 8, 677–687.
- Lechler, T.; Fuchs, E. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature 2005, 437, 275–280.
- Quyn, A.J.; Appleton, P.L.; Carey, F.A.; Steele, R.J.; Barker, N.; Clevers, H.; Ridgway, R.A.; Sansom, O.J.; Nathke, I.S. Spindle orientation bias in gut epithelial stem cell compartments is lost in precancerous tissue. Cell Stem Cell 2010, 6, 175–181.
- Cicalese, A.; Bonizzi, G.; Pasi, C.E.; Faretta, M.; Ronzoni, S.; Giulini, B.; Brisken, C.; Minucci, S.; Di Fiore, P.P.; Pelicci, P.G. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell 2009, 138, 1083–1095.
- Wu, M.; Kwon, H.Y.; Rattis, F.; Blum, J.; Zhao, C.; Ashkenazi, R.; Jackson, T.L.; Gaiano, N.; Oliver, T.; Reya, T. Imaging hematopoietic precursor division in real time. Cell Stem Cell 2007, 1, 541–554.
- Costa, M.R.; Wen, G.; Lepier, A.; Schroeder, T.; Gotz, M. Par-complex proteins promote proliferative progenitor divisions in the developing mouse cerebral cortex. Development 2008, 135, 11–22.
- Wang, X.; Tsai, J.W.; Imai, J.H.; Lian, W.N.; Vallee, R.B.; Shi, S.H. Asymmetric centrosome inheritance maintains neural progenitors in the neocortex. Nature 2009, 461, 947–955.
- Matthay, K.K.; Villablanca, J.G.; Seeger, R.C.; Stram, D.O.; Harris, R.E.; Ramsay, N.K.; Swift, P.; Shimada, H.; Black, C.T.; Brodeur, G.M.; et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N. Engl. J. Med. 1999, 341, 1165–1173.
- Izumi, H.; Kaneko, Y.; Nakagawara, A. The Role of MYCN in Symmetric vs. Asymmetric Cell Division of Human Neuroblastoma Cells. Front. Oncol. 2020, 10, 570815.
- Izumi, H.; Kaneko, Y. Evidence of asymmetric cell division and centrosome inheritance in human neuroblastoma cells. Proc. Natl. Acad. Sci. USA 2012, 109, 18048–18053.
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 2014, 4, a014357.
- Knoepfler, P.S.; Cheng, P.F.; Eisenman, R.N. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002, 16, 2699–2712.
- Giannini, G.; Cerignoli, F.; Mellone, M.; Massimi, I.; Ambrosi, C.; Rinaldi, C.; Dominici, C.; Frati, L.; Screpanti, I.; Gulino, A. High mobility group A1 is a molecular target for MYCN in human neuroblastoma. Cancer Res. 2005, 65, 8308–8316.
- Guo, Y.F.; Duan, J.J.; Wang, J.; Li, L.; Wang, D.; Liu, X.Z.; Yang, J.; Zhang, H.R.; Lv, J.; Yang, Y.J.; et al. Inhibition of the ALDH18A1-MYCN positive feedback loop attenuates MYCN-amplified neuroblastoma growth. Sci. Transl. Med. 2020, 12.
- Suenaga, Y.; Islam, S.M.; Alagu, J.; Kaneko, Y.; Kato, M.; Tanaka, Y.; Kawana, H.; Hossain, S.; Matsumoto, D.; Yamamoto, M.; et al. NCYM, a Cis-antisense gene of MYCN, encodes a de novo evolved protein that inhibits GSK3beta resulting in the stabilization of MYCN in human neuroblastomas. PLoS Genet 2014, 10, e1003996.
- Kaneko, Y.; Suenaga, Y.; Islam, S.M.; Matsumoto, D.; Nakamura, Y.; Ohira, M.; Yokoi, S.; Nakagawara, A. Functional interplay between MYCN, NCYM, and OCT4 promotes aggressiveness of human neuroblastomas. Cancer Sci. 2015, 106, 840–847.
- Puca, F.; Tosti, N.; Federico, A.; Kuzay, Y.; Pepe, A.; Morlando, S.; Savarese, T.; D’Alessio, F.; Colamaio, M.; Sarnataro, D.; et al. HMGA1 negatively regulates NUMB expression at transcriptional and post transcriptional level in glioblastoma stem cells. Cell Cycle 2019, 18, 1446–1457.
- Puca, F.; Colamaio, M.; Federico, A.; Gemei, M.; Tosti, N.; Bastos, A.U.; Del Vecchio, L.; Pece, S.; Battista, S.; Fusco, A. HMGA1 silencing restores normal stem cell characteristics in colon cancer stem cells by increasing p53 levels. Oncotarget 2014, 5, 3234–3245.
- Colaluca, I.N.; Tosoni, D.; Nuciforo, P.; Senic-Matuglia, F.; Galimberti, V.; Viale, G.; Pece, S.; Di Fiore, P.P. NUMB controls p53 tumour suppressor activity. Nature 2008, 451, 76–80.
- Izumi, H.; Kaneko, Y. Trim32 facilitates degradation of MYCN on spindle poles and induces asymmetric cell division in human neuroblastoma cells. Cancer Res. 2014.
- Betschinger, J.; Mechtler, K.; Knoblich, J.A. Asymmetric segregation of the tumor suppressor brat regulates self-renewal in Drosophila neural stem cells. Cell 2006, 124, 1241–1253.
- Schwamborn, J.C.; Berezikov, E.; Knoblich, J.A. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell 2009, 136, 913–925.