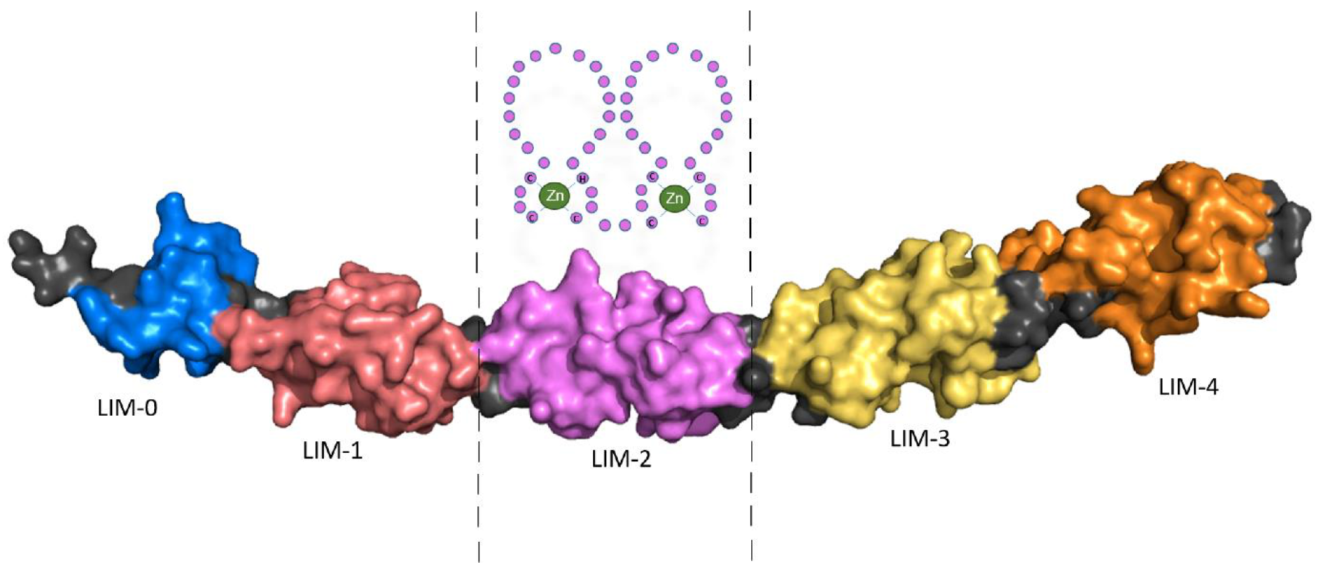
Four and a half LIM domains 2 (FHL2) was originally described as ‘Down-regulated in Rhabdomyosarcoma LIM protein’ (DRAL) and is composed of LIM domains that are named after their initial discovery in the proteins Lin11, Isl-1 and Mec-3. FHL2 consists of four and a half LIM domains, each composed of two zinc fingers, except the first ‘half’ LIM-domain which has only one. While the structure of the four and a half LIM domains had been uncovered previously using Nuclear Magnetic Resonance (NMR) spectroscopy, the complete FHL2 protein structure is unknown; however, it has recently been predicted using the protein structure neural network AlphaFold.
- obesity and related metabolic diseases
- epigenetic marks
- transcription factors
- metabolic diseases
- gene expression
1. Introduction
Schematic representation of LIM domain 2 (LIM-2) composed of two zinc fingers is shown in Figure 1.
2. Methylation of the FHL2 Gene
2.1. Hyper-Methylation of FHL2 in Aging
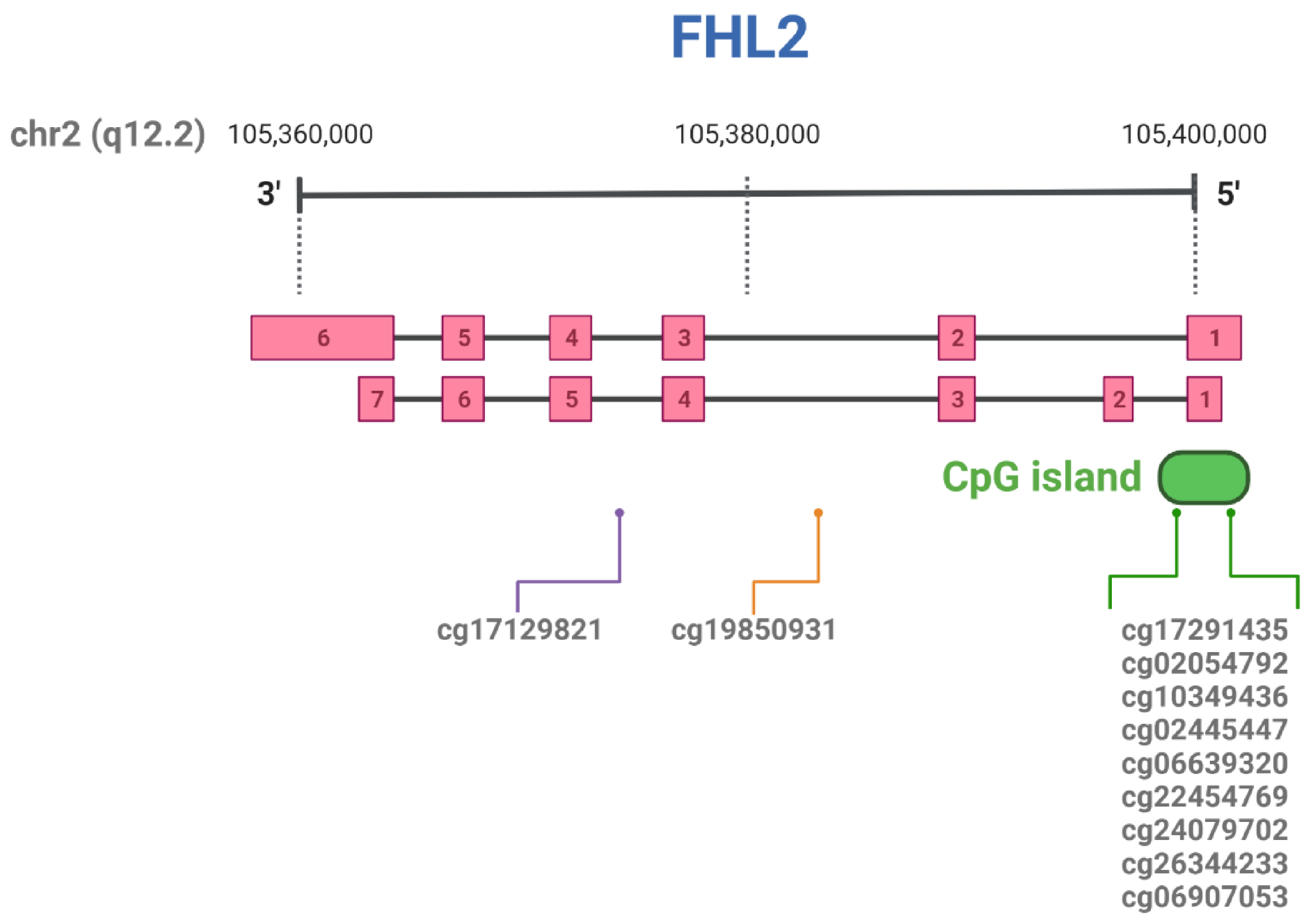
| CpG ID | Position in Chromosome |
Tissue | References |
|---|---|---|---|
| cg17129821 | chr2:105986385 a | Liver | [33] |
| cg19850931 | chr2:105993347 a | Whole blood | [29] |
| Adipose tissue | [29] | ||
| cg17291435 | chr2:106015527 a | Liver | [33] |
| cg02054792 | chr2:106014950 a | Liver | [33] |
| cg10349436 | chr2:106015079 a | Adipose tissue | [29] |
| cg02445447 | chr2:106015595 a | Liver | [33] |
| cg06639320 | chr2:106015740 a | Whole blood | [34][30][35][36][37] |
| Pancreatic islets | [34] | ||
| Leucocytes | [38] | ||
| Granulocytes | [39] | ||
| Liver | [33] | ||
| Lymphoblastoid line | [40] | ||
| Saliva | [22] | ||
| cg22454769 | chr2:106015768 a | Whole blood | [34][30][35][36][37] |
| Adipose tissue | [29] | ||
| Pancreatic islets | [34] | ||
| Leucocytes | [38] | ||
| Granulocytes | [39] | ||
| Liver | [33] | ||
| Skeletal muscle | [41] | ||
| cg24079702 | chr2:106015772 a | Whole blood | [34][30][35][36][37] |
| Pancreatic islets | [34] | ||
| Leucocytes | [38] | ||
| Granulocytes | [39] | ||
| Liver | [33] | ||
| cg26344233 | chr2:106015818 a | Whole blood | [29] |
| Adipose tissue | [29] | ||
| cg06907053 | chr2:106015870 a | Whole blood | [29] |
| Adipose tissue | [29] | ||
| Liver | [33] | ||
| Not specified (8 CpG sites) | chr2: 106015678–106016008 a | Teeth | [42] |
| Not specified (12 CpG sites) | chr2:105399282–105399340 b | Whole blood | [43] |
| Not specified | chr2:105399282 b | Whole blood | [44] |
| Not specified | chr2:105399291 b | Whole blood | [31] |
2.2. Hyper-Methylation of FHL2 in Metabolism
3. Concluding Remarks and Perspective
This entry is adapted from the peer-reviewed paper 10.3390/cells10102611
References
- Orii, N.; Ganapathiraju, M.K. Wiki-Pi: A Web-Server of Annotated Human Protein-Protein Interactions to Aid in Discovery of Protein Function. PLoS ONE 2012, 7, e49029.
- Tran, M.K.; Kurakula, K.; Koenis, D.S.; de Vries, C.J.M. Protein-protein interactions of the LIM-only protein FHL2 and functional implication of the interactions relevant in cardiovascular disease. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 219–228.
- Chen, C.Y.; Tsai, H.Y.; Tsai, S.H.; Chu, P.H.; Huang, P.H.; Chen, J.W.; Lin, S.J. Deletion of the FHL2 gene attenuates intima-media thickening in a partially ligated carotid artery ligated mouse model. J. Cell. Mol. Med. 2020, 24, 160–173.
- Liang, Y.; Bradford, W.H.; Zhang, J.; Sheikh, F. Four and a half LIM domain protein signaling and cardiomyopathy. Biophys. Rev. 2018, 10, 1073–1085.
- Liu, Z.; Han, S.; Wang, Y.; Cui, C.; Zhu, Q.; Jiang, X.; Yang, C.; Du, H.; Yu, C.; Li, Q.; et al. The LIM-only protein FHL2 is involved in autophagy to regulate the development of skeletal muscle cell. Int. J. Biol. Sci. 2019, 15, 838–846.
- van de Pol, V.; Vos, M.; DeRuiter, M.C.; Goumans, M.J.; de Vries, C.J.M.; Kurakula, K. LIM-only protein FHL2 attenuates inflammation in vascular smooth muscle cells through inhibition of the NFκB pathway. Vascul. Pharmacol. 2020, 125–126, 106634.
- Jin, X.; Jiao, X.; Jiao, J.; Zhang, T.; Cui, B. Increased expression of FHL2 promotes tumorigenesis in cervical cancer and is correlated with poor prognosis. Gene 2018, 669, 99–106.
- Sun, L.; Yu, S.; Xu, H.; Zheng, Y.; Lin, J.; Wu, M.; Wang, J.; Wang, A.; Lan, Q.; Furnari, F.; et al. FHL2 interacts with EGFR to promote glioblastoma growth. Oncogene 2018, 37, 1386–1398.
- Verset, L.; Feys, L.; Trépant, A.L.; De Wever, O.; Demetter, P. FHL2: A scaffold protein of carcinogenesis, tumour-stroma interactions and treatment response. Histol. Histopathol. 2016, 31, 469–478.
- Wang, C.; Lv, X.; He, C.; Davis, J.S.; Wang, C.; Hua, G. Four and a half lim domains 2 (FHL2) contribute to the epithelial ovarian cancer carcinogenesis. Int. J. Mol. Sci. 2020, 21, 7751.
- Chu, P.-H.; Bardwell, W.M.; Gu, Y.; Ross, J.; Chen, J. FHL2 (SLIM3) Is Not Essential for Cardiac Development and Function. Mol. Cell. Biol. 2000, 20, 7460–7462.
- Neuman, N.A.; Ma, S.; Schnitzler, G.R.; Zhu, Y.; Lagna, G.; Hata, A. The four-and-a-half LIM domain protein 2 regulates vascular smooth muscle phenotype and vascular tone. J. Biol. Chem. 2009, 284, 13202–13212.
- Purcell, N.H.; Darwis, D.; Bueno, O.F.; Müller, J.M.; Schüle, R.; Molkentin, J.D. Extracellular Signal-Regulated Kinase 2 Interacts with and Is Negatively Regulated by the LIM-Only Protein FHL2 in Cardiomyocytes. Mol. Cell. Biol. 2004, 24, 1081–1095.
- Clemente-Olivo, M.P.; Habibe, J.J.; Vos, M.; Ottenhoff, R.; Jongejan, A.; Herrema, H.; Zelcer, N.; Kooijman, S.; Rensen, P.C.N.; van Raalte, D.H.; et al. Four-and-a-half LIM domain protein 2 (FHL2) deficiency protects mice from diet-induced obesity and high FHL2 expression marks human obesity. Metabolism 2021, 121, 154815.
- Ng, C.F.; Zhou, W.J.W.; Ng, P.K.S.; Li, M.S.; Ng, Y.K.; Lai, P.B.S.; Tsui, S.K.W. Characterization of human FHL2 transcript variants and gene expression regulation in hepatocellular carcinoma. Gene 2011, 481, 41–47.
- Bai, S.; Zha, J.; Zhao, H.; Ross, F.P.; Teitelbaum, S.L. Tumor necrosis factor receptor-associated factor 6 is an intranuclear transcriptional coactivator in osteoclasts. J. Biol. Chem. 2008, 283, 30861–30867.
- Lv, H.; Zhang, M.; Shang, Z.; Li, J.; Zhang, S.; Lian, D.; Zhang, R. Genome-wide haplotype association study identify the FGFR2 gene as a risk gene for Acute Myeloid Leukemia. Oncotarget 2017, 8, 7891–7899.
- McKay, J.D.; Hung, R.J.; Han, Y.; Zong, X.; Carreras-Torres, R.; Christiani, D.C.; Caporaso, N.E.; Johansson, M.; Xiao, X.; Li, Y.; et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet. 2017, 49, 1126–1132.
- Howard, D.M.; Adams, M.J.; Shirali, M.; Clarke, T.K.; Marioni, R.E.; Davies, G.; Coleman, J.R.I.; Alloza, C.; Shen, X.; Barbu, M.C.; et al. Genome-wide association study of depression phenotypes in UK Biobank identifies variants in excitatory synaptic pathways. Nat. Commun. 2018, 9, 1–10.
- Palumbo, D.; Affinito, O.; Monticelli, A.; Cocozza, S. DNA Methylation variability among individuals is related to CpGs cluster density and evolutionary signatures. BMC Genomics 2018, 19, 229.
- Ciccarone, F.; Tagliatesta, S.; Caiafa, P.; Zampieri, M. DNA methylation dynamics in aging: How far are we from understanding the mechanisms? Mech. Ageing Dev. 2018, 174, 3–17.
- Jung, S.E.; Lim, S.M.; Hong, S.R.; Lee, E.H.; Shin, K.J.; Lee, H.Y. DNA methylation of the ELOVL2, FHL2, KLF14, C1orf132/MIR29B2C, and TRIM59 genes for age prediction from blood, saliva, and buccal swab samples. Forensic Sci. Int. Genet. 2019, 38, 1–8.
- Lee, H.Y.; Hong, S.R.; Lee, J.E.; Hwang, I.K.; Kim, N.Y.; Lee, J.M.; Fleckhaus, J.; Jung, S.E.; Lee, Y.H. Epigenetic age signatures in bones. Forensic Sci. Int. Genet. 2020, 46, 102261.
- Zbieć-Piekarska, R.; Spólnicka, M.; Kupiec, T.; Parys-Proszek, A.; Makowska, Z.; Pałeczka, A.; Kucharczyk, K.; Płoski, R.; Branicki, W. Development of a forensically useful age prediction method based on DNA methylation analysis. Forensic Sci. Int. Genet. 2015, 17, 173–179.
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492.
- Smith, J.; Sen, S.; Weeks, R.J.; Eccles, M.R.; Chatterjee, A. Promoter DNA Hypermethylation and Paradoxical Gene Activation. Trends Cancer 2020, 6, 392–406.
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115.
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.V.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2013, 49, 359–367.
- Rönn, T.; Volkov, P.; Gillberg, L.; Kokosar, M.; Perfilyev, A.; Jacobsen, A.L.; Jørgensen, S.W.; Brøns, C.; Jansson, P.A.; Eriksson, K.F.; et al. Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum. Mol. Genet. 2015, 24, 3792–3813.
- Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Gori, D.; Giuliani, C.; Mari, D.; Di Blasio, A.M.; Gentilini, D.; Vitale, G.; Collino, S.; et al. Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell 2012, 11, 1132–1134.
- Correia Dias, H.; Cunha, E.; Corte Real, F.; Manco, L. Age prediction in living: Forensic epigenetic age estimation based on blood samples. Leg. Med. 2020, 47, 101763.
- Steegenga, W.T.; Boekschoten, M.V.; Lute, C.; Hooiveld, G.J.; De Groot, P.J.; Morris, T.J.; Teschendorff, A.E.; Butcher, L.M.; Beck, S.; Müller, M. Genome-wide age-related changes in DNA methylation and gene expression in human PBMCs. Age (Omaha) 2014, 36, 1523–1540.
- Bysani, M.; Perfilyev, A.; De Mello, V.D.; Rönn, T.; Nilsson, E.; Pihlajamäki, J.; Ling, C. Epigenetic alterations in blood mirror age-associated DNA methylation and gene expression changes in human liver. Epigenomics 2017, 9, 105–122.
- Bacos, K.; Gillberg, L.; Volkov, P.; Olsson, A.H.; Hansen, T.; Pedersen, O.; Gjesing, A.P.; Eiberg, H.; Tuomi, T.; Almgren, P.; et al. Blood-based biomarkers of age-associated epigenetic changes in human islets associate with insulin secretion and diabetes. Nat. Commun. 2016, 7, 11089.
- Kananen, L.; Marttila, S.; Nevalainen, T.; Jylhävä, J.; Mononen, N.; Kähönen, M.; Raitakari, O.T.; Lehtimäki, T.; Hurme, M. Aging-associated DNA methylation changes in middle-aged individuals: The Young Finns study. BMC Genom. 2016, 17, 1–12.
- Freire-Aradas, A.; Phillips, C.; Mosquera-Miguel, A.; Girón-Santamaría, L.; Gómez-Tato, A.; Casares De Cal, M.; Álvarez-Dios, J.; Ansede-Bermejo, J.; Torres-Español, M.; Schneider, P.M.; et al. Development of a methylation marker set for forensic age estimation using analysis of public methylation data and the Agena Bioscience EpiTYPER system. Forensic Sci. Int. Genet. 2016, 24, 65–74.
- Cho, S.; Jung, S.E.; Hong, S.R.; Lee, E.H.; Lee, J.H.; Lee, S.D.; Lee, H.Y. Independent validation of DNA-based approaches for age prediction in blood. Forensic Sci. Int. Genet. 2017, 29, 250–256.
- Johansson, Å.; Enroth, S.; Gyllensten, U. Continuous Aging of the Human DNA Methylome Throughout the Human Lifespan. PLoS ONE 2013, 8, e67378.
- Bacalini, M.G.; Deelen, J.; Pirazzini, C.; De Cecco, M.; Giuliani, C.; Lanzarini, C.; Ravaioli, F.; Marasco, E.; Van Heemst, D.; Suchiman, H.E.D.; et al. Systemic Age-Associated DNA Hypermethylation of ELOVL2 Gene: In Vivo and in Vitro Evidences of a Cell Replication Process. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2017, 72, 1015–1023.
- Taniguchi, I.; Iwaya, C.; Ohnaka, K.; Shibata, H.; Yamamoto, K. Genome-wide DNA methylation analysis reveals hypomethylation in the low-CpG promoter regions in lymphoblastoid cell lines. Hum. Genom. 2017, 11, 8.
- Turner, D.C.; Gorski, P.P.; Maasar, M.F.; Seaborne, R.A.; Baumert, P.; Brown, A.D.; Kitchen, M.O.; Erskine, R.M.; Dos-Remedios, I.; Voisin, S.; et al. DNA methylation across the genome in aged human skeletal muscle tissue and muscle-derived cells: The role of HOX genes and physical activity. Sci. Rep. 2020, 10, 1–19.
- Giuliani, C.; Cilli, E.; Bacalini, M.G.; Pirazzini, C.; Sazzini, M.; Gruppioni, G.; Franceschi, C.; Garagnani, P.; Luiselli, D. Inferring chronological age from DNA methylation patterns of human teeth. Am. J. Phys. Anthropol. 2016, 159, 585–595.
- Correia Dias, H.; Cordeiro, C.; Corte Real, F.; Cunha, E.; Manco, L. Age Estimation Based on DNA Methylation Using Blood Samples From Deceased Individuals. J. Forensic Sci. 2020, 65, 465–470.
- Dias, H.C.; Cordeiro, C.; Pereira, J.; Pinto, C.; Real, F.C.; Cunha, E.; Manco, L. DNA methylation age estimation in blood samples of living and deceased individuals using a multiplex SNaPshot assay. Forensic Sci. Int. 2020, 311, 110267.
- Wan, J.; Oliver, V.F.; Wang, G.; Zhu, H.; Zack, D.J.; Merbs, S.L.; Qian, J. Characterization of tissue-specific differential DNA methylation suggests distinct modes of positive and negative gene expression regulation. BMC Genom. 2015, 16, 49.