Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Mitochondrial dysfunction and stem cell exhaustion are two hallmarks of aging. In the hematopoietic system, aging is linked to imbalanced immune response and reduced regenerative capacity in hematopoietic stem cells (HSCs), as well as an increased predisposition to a spectrum of diseases, including myelodysplastic syndrome and acute myeloid leukemia.
- hematopoiesis
- hematopoietic stem cell
- aging
- mitochondrial metabolism
- stem cell exhaustion
- ROS
- inflammation
1. Introduction
Aging is a time-dependent degenerative process that affects all living organisms. Since the aging population is inexorably growing, there is an imminent need to develop new therapeutic strategies for ameliorating the age-related changes and/or disorders, and first among these is hematopoietic aging. The milestone review paper of Reference [1] has categorized the cellular and molecular hallmarks of this type of aging, which include both mitochondrial dysfunction and stem cell exhaustion, the two main topics of this review.
Mitochondria were described as contributing to aging and degeneration as early as already in the 1920s, when the “rate of living hypothesis” proposed that metabolic rates inversely correlate with organismal lifespan [2]. Many researchers have since described the links between mitochondrial biology and aging [3,4,5,6]. Stem cell exhaustion refers to an impaired functionality of stem cells, which cannot maintain in the tissue in which they reside. In particular, aging of the hematopoietic system displays decreased immune response, declining immuno-competence, increased autoimmunity, diminished stress response, late-onset anemia, reduced regenerative capacity, and increased predisposition to a spectrum of diseases, including myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) [7,8,9,10].
Several distinct features characterize aged hematopoietic stem cells (HSCs) (Figure 1) [11]. For example, it has been shown that phenotypic HSCs in the bone marrow increase in frequency with age, while losing their functionality. Multiple studies have demonstrated a differentiation bias toward the myeloid lineage; the aged murine hematopoietic system is impaired in supporting leukocyte numbers, erythropoiesis, and both B- and T-lymphoid cells in peripheral blood, while the numbers of myeloid cells are increased [12]. These myeloid-biased HSCs express high levels of CD150 (signaling lymphocyte activation molecule, or SLAMF1) and CD41 (integrin alpha 2, or Itga2b) proteins, which have been used to identify the HSC clonal subtypes responsible for hematopoietic aging [13,14]. As expected, stemness decreases in aged HSCs, which show reduced in vivo repopulation capacity, as determined by serial transplantation assays [15], along with a 3-fold lower efficiency in bone marrow homing after transplantation (Figure 1) [16].
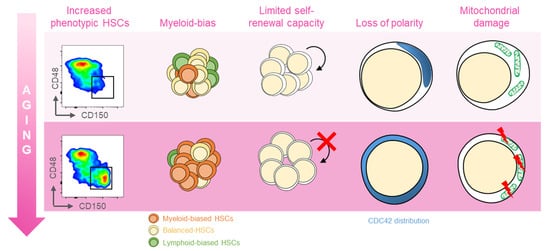
Figure 1. Features of aged HSCs. Upon aging, hematopoietic stem cells (HSCs) acquire phenotypical and functional peculiar properties. Flow cytometry analysis of the murine bone marrow shows an increase in the phenotypic HSCs (defined as c-Kit+Sca-1+Lin−CD135−CD48−CD150+) in old (18 months old, bottom panel) mice (far left). The HSC pool includes balanced HSC, which in equal proportion differentiate in myeloid and lymphoid lineage (yellow, left); with age, the myeloid-biased differentiation prevails at the expense of lymphoid cells (left). The self-renewal capacity typical of HSCs is reduced upon aging (middle). Cytoskeletal polarity detected by CDC42 localization is lost in old HSCs where CDC42 expression is homogeneously distributed (right). Aged HSCs display the mitochondrial damage with the altered metabolism (far right).
Another key feature of aged HSCs is the loss of cell polarity. Young and aged long-term (LT) HSCs exhibit different distribution patterns of CDC42, Tubulin, and AcH4K16, which is believed to be caused by the elevated activity of CDC42, and is linked to age-associated changes in self-renewal and differentiation capacity [12]. Additionally, HSC aging results from cumulative cellular and genomic damage, which leads to permanent cell-cycle arrest, apoptosis, or senescence [15,17,18].
Although historically DNA damage was thought to be the main cause of HSC aging, many new findings have defined an increasing number of biological processes that intrinsically change with age in HSCs. These include epigenetics, chromatin architecture, autophagy, proteostasis, and metabolic changes [19].
Since aged HSCs generally exhibit enhanced mitochondrial oxidative phosphorylation (OXPHOS) and increased production of reactive oxygen species (ROS), it has often been proposed that mitochondria play a direct role in compromising HSC functions [20,21] (Figure 2).
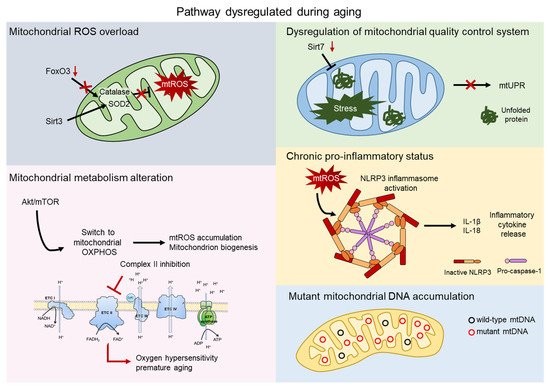
Figure 2. Mitochondrial contribution to aging. Aging process shows dysregulation of several pathway involving the direct contribution of mitochondria. Mitochondrial reactive oxygen species (mtROS) accumulate after the impairment of the major antioxidant defense systems, consist of Superoxide dismutase 2 (SOD2) and Catalase, caused by the aged-related loss of FOXO3 and SIRT3 (grey). AKT/mTOR signaling is a key regulator of mitochondrial metabolism and, upon aging, its imbalance causes mtROS accumulation, mitochondrial biogenesis, and an altered metabolism. Inhibition of complex II of the electron transport chain (ETC) leads to oxygen hypersensitivity and premature aging of HSCs (pink). The mitochondrial unfolded protein response system (mtUPR) is affected by the aged-related loss of SIRT7, leading to accumulation of unfolded protein and mitochondrial stress (green). Accumulation of mtROS activates the NOD-, LRR-, and pyrin domain-containing 3 (NLRP3) inflammasome, which triggers the release of inflammatory cytokines, such as IL-1β and IL-18 (orange). Mitochondrial dysfunctions include accumulation of mtDNA mutations in aged HSCs (blue).
2. Mitochondrial ROS
Since the frequency of HSCs with low levels of ROS decreases with age, ROS generation/accumulation can be considered a distinctive characteristic of aging [22]. Proper levels of ROS are important mediators of various signal transduction pathways. However, increased levels of ROS affect HSCs’s lifespan [20], self-renewal [23,24], and differentiation [25,26]. ROS contribute to HSC aging and senescence, and excessive ROS generation induces apoptotic cell death in HSCs [20,23,27,28]. Increase in ROS levels in adult HSC have similarities with the aging phenotypes, such as myeloid lineage skewing and defective long-term repopulation activity [29,30]. On the other hand, very low levels of intracellular ROS in HSCs are essential to maintaining HSCs quiescence [22]. Mitochondria produce around 90% of cellular ROS, and the impairment of mitochondrial function, for example, by the loss of the Polycomb repressor BMI1, causes a major increase in intracellular ROS [31]. Aging of mitochondria leads to an overload of ROS, which further damage the mitochondria, resulting in perpetual cell cycling [32]. Evidence for this has come from studies of the effects of FOXO transcription factors, key players in the oxidative stress response, on HSC fitness. Genetic variation within the FOXO3 gene is associated with human longevity and aging phenotypes [33]. Mice carrying triple conditional deletions of FOXO1, FOXO3a, and FOXO4 genes in the adult hematopoietic system exhibited myeloid lineage expansion, lymphoid developmental abnormalities, and a decreased long-term repopulation ability in vivo while increased ROS levels [30]. Another study from Dr. Tosho Suda’s group also demonstrated that FOXO3a-deleted HSCs can neither maintain quiescence nor support long-term reconstitution of hematopoiesis in vivo [34]. FOXO3a deficiency increased levels of ROS and downregulated several cyclin-dependent kinases inhibitors, resulting in the exit of HSCs from quiescence. This boosted sensitivity to cell-cycle-specific myelotoxic injury, and loss of self-renewal capacity during aging [34]. FOXO3a knockout HSCs also showed lower expression of mitochondrial Superoxide dismutase 2 (SOD2) and Catalase, two FOXO targets involved in ROS detoxification [35,36]. Further, FOXO3 has been shown to be crucial for the regulation of mitochondrial respiration in HSCs, which, under disrupted conditions, generate more ROS [29]. This strengthens the hypothesis that FOXO3a deficiency causes HSCs cell cycle abnormalities via mitochondrial ROS dysregulation.
Mitochondrial ROS levels and the related signaling pathways, thus, represent a major player in regulating the long-term self-renewal, activation, proliferation, differentiation, and aging of HSCs. Similar roles could also be played by extrinsic factors and the surrounding microenvironment, which can have a direct impact on ROS levels and the signaling pathways regulating HSCs homeostasis [10].
ROS are produced as by-products of mitochondrial respiration; their production is increased, and they are accumulated when mitochondrial respiration is altered. The major ROS source is the mitochondrial electron transport Chain (ETC), which is widely targeted by mitochondrial DNA (mtDNA) mutations [37], as described in detail in the following dedicated paragraphs.
3. Mitochondrial Metabolism
Despite the high preference of mitochondria for glycolysis, recent studies have also highlighted the importance of mitochondrial respiration to HSC for proliferation and maintenance [48,49,50]. We have recently shown that HSCs have a relatively high number of mitochondria, which are not completely inactive [51]. Indeed, mitochondrial membrane potential (ΔΨmt) is high in HSCs, although ATP production or intracellular ROS levels are low [52,53]. The higher complex II: complex V ratio gives rise to high ΔΨmt in HSCs due to limited coupling of the electron transport chain (ETC), which supports the idea that mitochondrial complex II is pivotal for both HSC maintenance and the prevention of the aging process [53]. Indeed, inhibition of complex II reduces the in vitro colony-replating capacity of HSCs [53], and genetic mutation of mev-1, a subunit of the succinate dehydrogenase cytochrome b enzyme, which is a component of complex II, leads to oxygen hypersensitivity and premature aging of HSCs [54]. Studies of C. elegans uncovered that the mutated or silenced components of ETC or the ATP synthase can markedly extend [55,56,57] or reduce lifespan [58]. Although these varying experimental results must eventually be resolved, it is clear that imbalances in ETC activity are closely linked to the overall survival of the organism. Interestingly, a recent paper showed that ΔΨmt is a source of heterogeneity in old HSCs, with a prevalent fraction of low ΔΨmt in aged HSCs. Enhancement of ΔΨmt by mitoquinol (Mito-Q), a mitochondrial-targeted coenzyme-Q10 [59], successfully increased ΔΨmt of old HSCs and ameliorated or prevented onset of aging phenotypes [60].
HSCs are mainly dormant but can become highly active on demand, either to maintain hematopoietic homeostasis by replenishing matured/immature hematopoietic cells, or to respond to situations of emergency, such as infection or blood loss [61]. This shift requires a metabolic switch from glycolysis to mitochondrial oxidative phosphorylation, which is precisely regulated by various signaling pathways. The mammalian TOR (mTOR) pathway is a key regulator of cellular and mitochondrial metabolism. mTOR directly controls the mitochondrial oxidative function through a YY1–PGC-1α (peroxisome proliferator-activated receptor gamma coactivator 1-alpha) transcriptional complex [62]. Defects of Tuberous sclerosis complex subunit 1 (TSC1), the major negative regulator for mTOR [63], lead to increased mitochondrion biogenesis and accumulation of ROS. Blockade of ROS activity in vivo restores these HSC defects, demonstrating that the TSC-mTOR pathway controls the quiescence and on-demand functions of HSCs by repressing ROS production [64,65].
HSCs exhibit low AKT/mTOR activity, but, upon stress, upregulation of this pathway drives dormant HSCs toward activation [64,66]. Interestingly, the dysregulation of AKT/mTOR signaling correlates with the aging process in HSCs [67]. Experimental evidence has shown that mTOR activation is involved in HSC aging, as well as that rapamycin treatment restores HSC potential and prolongs the lifespan of mice [68]. mTOR activity is higher in HSCs from elder mice than younger mice, and mTOR activation, through conditional deletion of TSC1 in the HSCs of young mice, mimics the phenotype of HSCs from aged mice; similarly, in older mice, rapamycin restores the self-renewal capacity of HSCs and, importantly, correlates with increased life span [68].
ASXL1 is frequently mutated in age-related clonal hematopoiesis. Its mutation activates the AKT/mTOR pathway, causing aberrant cell cycle progression in the HSC compartment and provoking HSC dysfunction. This is associated with mitochondrial activation, elevated ROS levels, and increased DNA damage, leading to age-associated phenotypes, such as myeloid-biased differentiation, hypocellular bone marrow, and increased frequency, of phenotypic LT-HSCs. Inhibition of the AKT/mTOR pathway can partially rescue these phenotypes, suggesting its involvement in the enhanced aging of the hematopoietic system [69].
A similar phenotype is observed in wild-type p53-induced phosphatase 1 (WIP1), which is highly expressed in HSCs but decreases with age. WIP1-deficient (WIP1−/−) mice exhibit multiple aging-like phenotypes in HSCs, including declines in reconstitution ability and HSC expansion. Mechanistically, their impaired regenerative capacity is due to a p53-mediated differentiation defect, whereas increasing numbers of WIP1−/− HSCs are associated with mTOR-mediated cell cycle progression of HSCs [70]. Notably, experimental results have shown that aged HSCs have higher mTOR [71] activity levels, as well as that its inhibitor rapamycin can restore the self-renewal of aged HSCs, an effect which can be translated to human HSCs [72].
Recent advances have demonstrated that epigenetic, transcriptional, and post-transcriptional mechanisms also control the quiescence of HSCs, which are maintained in a paused state that allows for rapid activation [73]. Mitochondrial activity modifies the epigenetic state of cells affecting their aging process [74]. Citric acid, generated by the tricarboxylic acid (TCA) cycle in the mitochondria, modulates histone acetylation and gene expression through its conversion to acetyl-CoA [74]. Mitochondrial fatty acid oxidation (FAO) also generates acetyl-CoA for histone modification in HSCs [75].
Sirtuins are a family of protein deacetylases, which regulate the mitochondrial metabolic checkpoint in stem cells, and they are key regulators of stem cell aging [21,76]. SIRT3 plays a critical role in the mitochondria, where it deacetylates two critical lysine residues on SOD2 to promote the antioxidative activity. Brown and colleagues have demonstrated that SIRT3 is highly enriched in HSCs, as well as is suppressed with aging [77]. Although SIRT3 has no effect on HSCs maintenance or tissue homeostasis at a young age under homeostatic conditions, it is essential under stress or in old age. Indeed, SIRT3 loss induces HSC quiescence and compromises regenerative capacity in old mice [77].
SIRT1 is a key regulator of HSCs self-renewal and lineage specification under homeostasis. Interestingly, Ghaffari’s group has shown that loss of SIRT1 causes anemia and myeloid expansion at the expense of the lymphoid compartment, overlapping features with aged HSCs. SIRT1 plays a role in HSCs homeostasis by targeting FOXO3, a longevity transcription factor and mitochondrial ROS regulator [78].
Another key regulator of metabolism is nicotinamide adenine dinucleotide (NAD+). Decreased levels of NAD+ are associated with cancer, metabolic disorders, and physiological and accelerated aging processes [79,80,81]. Supplementation of nicotinamide riboside (NR), a NAD+ precursor, significantly improved lifespan and health span in model of aged-related disease, such as ataxia–telangiectasia mutation (ATM), thanks to the improvement of both DNA damage repair and mitophagy [71,82]. Murine models of ATM loss show defects in DNA damage repair associated with mitochondrial dysfunction [83] and loss of hematopoietic stem cell (HSC) potential [23]. NR treatment caused significant alterations in lineage commitment of HSCs with enhanced lymphoid potential [84].
This entry is adapted from the peer-reviewed paper 10.3390/ijms222011117
This entry is offline, you can click here to edit this entry!