Oxysterols are oxidized derivatives of cholesterol produced by enzymatic activity or non-enzymatic pathways (auto-oxidation). The oxidation processes lead to the synthesis of about 60 different oxysterols. Several oxysterols have physiological, pathophysiological, and pharmacological activities. The effects of oxysterols on cell death processes, especially apoptosis, autophagy, necrosis, and oxiapoptophagy (a complex mode of cell death characterized by ROS overproduction (“oxi-”), apoptosis induction, (“-apopto”), and autophagy (“-phagy”)), as well as their action on cell proliferation, are reviewed here. These effects, also observed in several cancer cell lines, could potentially be useful in cancer treatment. The effects of oxysterols on cell differentiation are also described. Among them, the properties of stimulating the osteogenic differentiation of mesenchymal stem cells while inhibiting adipogenic differentiation may be useful in regenerative medicine.
- apoptosis
- autophagy
- cell death
- differentiation
- oxysterol
- Oxiapoptophagy
1. Introduction
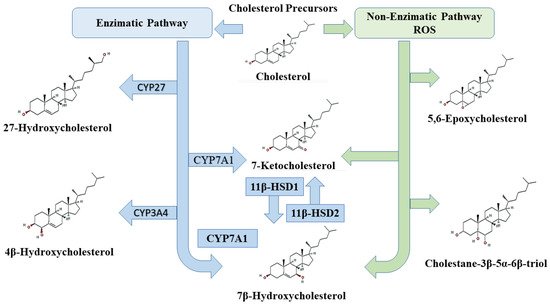
2. Oxysterols
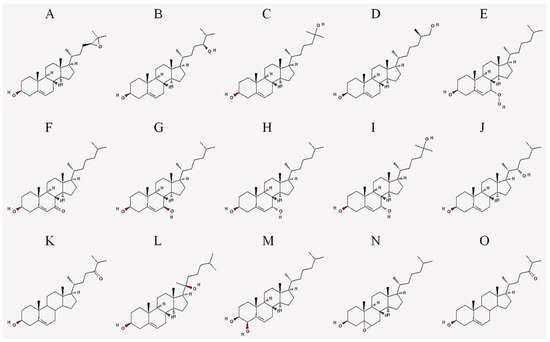
| Abbreviation | Common Name | IUPAC Name |
|---|---|---|
| 24,25-EC | 24(S),25-epoxycholesterol | (3S,8S,9S,10R,13R,14S,17R)-17-[(2R)-4-[(2S)-3,3-dimethyloxiran-2-yl]butan-2-yl]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-ol |
| 24-HC | 24(S)-hydroxycholesterol | (3S,8S,9S,10R,13R,14S,17R)-17-[(2R,5S)-5-hydroxy-6-methylheptan-2-yl]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-ol |
| 25-HC | 25-hydroxycholesterol | (3S,8S,9S,10R,13R,14S,17R)-17-[(2R)-6-hydroxy-6-methylheptan-2-yl]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-ol |
| 27-HC | 27-hydroxycholesterol | (3S,8S,9S,10R,13R,14S,17R)-17-[(2R,6R)-7-hydroxy-6-methylheptan-2-yl]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-ol |
| 7-OOHC | 7-hydroperoxycholesterol | (3S,8S,9S,10R,13R,14S,17R)-7-hydroperoxy-10,13-dimethyl-17-[(2R)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-ol |
| 7α-HC | 7α-hydroxycholesterol | (3S,7S,8S,9S,10R,13R,14S,17R)-10,13-dimethyl-17-[(2R)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthrene-3,7-diol |
| 7β-HC | 7β-hydroxycholesterol | (3S,4R,8S,9S,10R,13R,14S,17R)-10,13-dimethyl-17-[(2R)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthrene-3,4-diol |
| 7-KC | 7-ketocholesterol | (3S,8S,9S,10R,13R,14S,17R)-3-hydroxy-10,13-dimethyl-17-[(2R)-6-methylheptan-2-yl]-1,2,3,4,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-7-one |
| 7α,25-DHC | 7α,25-dihydroxycholesterol | (3S,7S,8S,9S,10R,13R,14S,17R)-17-[(2R)-6-hydroxy-6-methylheptan-2-yl]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthrene-3,7-diol |
| 7β,27-DHC | 7β,27-dihydroxycholesterol | (3S,7R,8S,9S,10R,13R,14S,17R)-17-[(2R)-7-hydroxy-6-methylheptan-2-yl]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthrene-3,7-diol |
| 22-HC | 22(S)-hydroxycholesterol | (3S,8S,9S,10R,13S,14S,17R)-17-[(2S,3S)-3-hydroxy-6-methylheptan-2-yl]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-ol |
| 20-HC | 20(S)-hydroxycholesterol | (3S,8S,9S,10R,13S,14S,17S)-17-[(2S)-2-hydroxy-6-methylheptan-2-yl]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-ol |
| 4β-HC | 4β-hydroxycholesterol | (3S,4R,8S,9S,10R,13R,14S,17R)-10,13-dimethyl-17-[(2R)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthrene-3,4-diol |
| 7,25-DHC | 7,25-dihydroxycholesterol | (3S,8S,9S,10R,13R,14S,17R)-17-[(2R)-6-hydroxy-6-methylheptan-2-yl]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthrene-3,7-diol |
| 5,6-EC | 5,6-epoxycholesterol | (1S,2R,5S,11S,12S,15R,16R)-2,16-dimethyl-15-[(2R)-6-methylheptan-2-yl]-8-oxapentacyclo[9.7.0.02,7.07,9.012,16]octadecan-5-ol |
| 24-OXO | 24-oxocholesterol | (6R)-6-[(3S,10R,13R,17R)-3-hydroxy-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-17-yl]-2-methylheptan-3-one |
3. Oxysterols and Cell Death
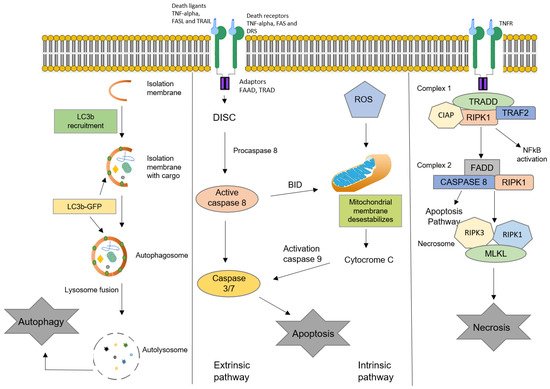
3.1. Apoptosis
3.2. Autophagy
3.3. Necrosis
This entry is adapted from the peer-reviewed paper 10.3390/cells10092301
References
- Danial, N.N.; Hockenbery, D.M. Chapter 18—Cell death. In Hematology, 7th ed.; Hoffman, R., Benz, E.J., Silberstein, L.E., Heslop, H.E., Weitz, J.I., Anastasi, J., Salama, M.E., Abutalib, S.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 186–196.
- Stenson, W.F.; Ciorba, M.A. Chapter 9—Cell death. In Physiology of the Gastrointestinal Tract, 6th ed.; Said, H.M., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 221–234.
- Rock, K.L.; Kono, H. The inflammatory response to cell death. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 99–126.
- Proskuryakov, S.Y.; Konoplyannikov, A.G.; Gabai, V.L. Necrosis: A specific form of programmed cell death? Exp. Cell. Res. 2003, 283, 1–16.
- Castelli, G.; Pelosi, E.; Testa, U. Emerging Therapies for Acute Myelogenus Leukemia Patients Targeting Apoptosis and Mitochondrial Metabolism. Cancers 2019, 11, 260.
- Mbaveng, A.; Bitchagno, G.T.; Kuete, V.; Tane, P.; Efferth, T. Cytotoxicity of ungeremine towards multi-factorial drug resistant cancer cells and induction of apoptosis, ferroptosis, necroptosis and autophagy. Phytomedicine 2019, 60, 152832.
- Duc, D.; Vigne, S.; Pot, C. Oxysterols in autoimmunity. Int. J. Mol. Sci. 2019, 20, 4522.
- Griffiths, W.J.; Wang, Y. Oxysterols as lipid mediators: Their biosynthetic genes, enzymes and metabolites. Prostaglandins Other Lipid Mediat. 2019, 147, 106381.
- Crick, P.J.; Yutuc, E.; Abdel-Khalik, J.; Saeed, A.; Betsholtz, C.; Genove, G.; Björkhem, I.; Wang, Y.; Griffiths, W.J. Formation and metabolism of oxysterols and cholestenoic acids found in the mouse circulation: Lessons learnt from deuterium-enrichment experiments and the CYP46A1 transgenic mouse. J. Steroid Biochem. Mol. Biol. 2019, 195, 105475.
- Levy, D.; Melo, T.; Ruiz, J.; Bydlowski, S.P. Oxysterols and mesenchymal stem cell biology. Chem. Phys. Lipids 2017, 207, 223–230.
- Zmysłowski, A.; Szterk, A. Oxysterols as a biomarker in diseases. Clin. Chim. Acta 2019, 491, 103–113.
- Ursan, R.; Odnoshivkina, U.G.; Petrov, A.M. Membrane cholesterol oxidation downregulates atrial β-adrenergic responses in ROS-dependent manner. Cell. Signal. 2020, 67, 109503.
- Mutemberezi, V.; Guillemot-Legris, O.; Muccioli, G.G. Oxysterols: From cholesterol metabolites to key mediators. Prog. Lipid Res. 2016, 64, 152–169.
- Riscal, R.; Skuli, N.; Simon, M.C. Even cancer cells watch their cholesterol! Mol. Cell 2019, 76, 220–231.
- Nury, T.; Zarrouk, A.; Ragot, K.; Debbabi, M.; Riedinger, J.-M.; Vejux, A.; Aubourg, P.; Lizard, G. 7-Ketocholesterol is increased in the plasma of X-ALD patients and induces peroxisomal modifications in microglial cells: Potential roles of 7-ketocholesterol in the pathophysiology of X-ALD. J. Steroid Biochem. Mol. Biol. 2017, 169, 123–136.
- Bischoff, P.; Holl, V.; Coelho, D.; Dufour, P.; Luu, B.; Weltin, D. Apoptosis at the Interface of Immunosuppressive and Anticancer Activities: The Examples of Two Classes of Chemical Inducers, Oxysterols and Alkylating Agents. Curr. Med. Chem. 2000, 7, 693–713.
- Chimento, A.; Casaburi, I.; Avena, P.; Trotta, F.; De Luca, A.; Rago, V.; Pezzi, V.; Sirianni, R. Cholesterol and Its Metabolites in Tumor Growth: Therapeutic Potential of Statins in Cancer Treatment. Front. Endocrinol. 2019, 9, 807.
- Rodriguez-Estrada, M.; Cardenia, V.; Poirot, M.; Iuliano, L.; Lizard, G. Oxysterols and sterols: From lipidomics to food sciences. Elsevier 2020, 196, 105515.
- Olkkonen, V.M.; Béaslas, O.; Nissilä, E. Oxysterols and Their Cellular Effectors. Biomolecules 2012, 2, 76–103.
- Oguro, H. The Roles of Cholesterol and Its Metabolites in Normal and Malignant Hematopoiesis. Front. Endocrinol. 2019, 10, 204.
- Griffiths, W.J.; Wang, Y. An update on oxysterol biochemistry: New discoveries in lipidomics. Biochem. Biophys. Res. Commun. 2018, 504, 617–622.
- Kloudova, A.; Guengerich, F.P.; Soucek, P. The role of oxysterols in human cancer. Trends Endocrinol. Metab. 2017, 28, 485–496.
- Hutchinson, S.A.; Lianto, P.; Moore, J.B.; Hughes, T.A.; Thorne, J.L. Phytosterols Inhibit Side-Chain Oxysterol Mediated Activation of LXR in Breast Cancer Cells. Int. J. Mol. Sci. 2019, 20, 3241.
- Vejux, A.; Namsi, A.; Nury, T.; Moreau, T.; Lizard, G. Biomarkers of Amyotrophic Lateral Sclerosis: Current Status and Interest of Oxysterols and Phytosterols. Front. Mol. Neurosci. 2018, 11, 12.
- Lin, Y.; Koppenol, W.P.; Knol, D.; Vermeer, M.A.; Hiemstra, H.; Friedrichs, S.; Lütjohann, D.; Trautwein, E.A.; Lin, K. Serum Concentration of Plant Sterol Oxidation Products (POP) Compared to Cholesterol Oxidation Products (COP) after Intake of Oxidized Plant Sterols: A Randomised, Placebo-Controlled, Double-Blind Dose‒Response Pilot Study. Nutrients 2019, 11, 2319.
- Brown, A.J.; Jessup, W. Oxysterols: Sources, cellular storage and metabolism, and new insights into their roles in cholesterol homeostasis. Mol. Asp. Med. 2009, 30, 111–122.
- Ito, H.; Matsuo, K.; Hosono, S.; Watanabe, M.; Kawase, T.; Suzuki, T.; Hirai, T.; Yatabe, Y.; Tanaka, H.; Tajima, K. Association between CYP7A1 and the risk of proximal colon cancer in Japanese. Int. J. Mol. Epidemiol. Genet. 2009, 1, 35–46.
- Srivastava, A.; Choudhuri, G.; Mittal, B. CYP7A1 (−204 A>C; rs3808607 and −469 T>C; rs3824260) promoter polymorphisms and risk of gallbladder cancer in North Indian population. Metabolism 2010, 59, 767–773.
- Zhou, L.-P.; Yao, F.; Luan, H.; Wang, Y.-L.; Dong, X.-H.; Zhou, W.-W.; Wang, Q.-H. CYP3A4* 1B polymorphism and cancer risk: A HuGE review and meta-analysis. Tumor Biol. 2013, 34, 649–660.
- Lu, L.; Zhao, G.; Ouellet, J.; Fan, Z.; Labrie, F.; Pelletier, G. Expression of 11β-hydroxysteroid dehydrogenase type 1 in breast cancer and adjacent non-malignant tissue. An immunocytochemical study. Pathol. Oncol. Res. 2011, 17, 627–632.
- Lipka, C.; Mankertz, J.; Fromm, M.; Lübbert, H.; Bühler, H.; Kühn, W.; Ragosch, V.; Hundertmark, S. Impairment of the antiproliferative effect of glucocorticosteroids by 11β-hydroxysteroid dehydrogenase type 2 overexpression in MCF-7 breast-cancer cells. Horm. Metab. Res. 2004, 36, 437–444.
- Luu, W.; Sharpe, L.J.; Capell-Hattam, I.; Gelissen, I.C.; Brown, A.J. Oxysterols: Old tale, new twists. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 447–467.
- Kanner, J.; Lapidot, T. The stomach as a bioreactor: Dietary lipid peroxidation in the gastric fluid and the effects of plant-derived antioxidants. Free Radic. Biol. Med. 2001, 31, 1388–1395.
- Dwyer, J.; Sever, N.; Carlson, M.; Nelson, S.F.; Beachy, P.A.; Parhami, F. Oxysterols Are Novel Activators of the Hedgehog Signaling Pathway in Pluripotent Mesenchymal Cells. J. Biol. Chem. 2007, 282, 8959–8968.
- Cheng, Y.; Kang, J.; Shih, Y.; Lo, Y.; Wang, C. Cholesterol-3-beta, 5-alpha, 6-beta-triol induced genotoxicity through reactive oxygen species formation. Food Chem. Toxicol. 2005, 43, 617–622.
- Aghaloo, T.L.; Amantea, C.M.; Cowan, C.M.; Richardson, J.A.; Wu, B.M.; Parhami, F.; Tetradis, S. Oxysterols enhance osteoblast differentiation in vitro and bone healing in vivo. J. Orthop. Res. 2007, 25, 1488–1497.
- Liu, X.; Tajima, N.; Taniguchi, M.; Kato, N. The enantiomer pair of 24S-and 24R-hydroxycholesterol differentially alter activity of large-conductance Ca2+-dependent K+ (slo1 BK) channel. Chirality 2020, 32, 223–230.
- Amantea, C.M.; Kim, W.-K.; Meliton, V.; Tetradis, S.; Parhami, F. Oxysterol-induced osteogenic differentiation of marrow stromal cells is regulated by Dkk-1 inhibitable and PI3-kinase mediated signaling. J. Cell. Biochem. 2008, 105, 424–436.
- Hong, W.; Guo, F.; Yang, M.; Xu, D.; Zhuang, Z.; Niu, B.; Bai, Q.; Li, X. Hydroxysteroid sulfotransferase 2B1 affects gastric epithelial function and carcinogenesis induced by a carcinogenic agent. Lipids Health Dis. 2019, 18, 1–14.
- Kloudova-Spalenkova, A.; Ueng, Y.-F.; Wei, S.; Kopeckova, K.; Guengerich, F.P.; Soucek, P. Plasma oxysterol levels in luminal subtype breast cancer patients are associated with clinical data. J. Steroid Biochem. Mol. Biol. 2019, 197, 105566.
- Vejux, A.; Abed-Vieillard, D.; Hajji, K.; Zarrouk, A.; Mackrill, J.J.; Ghosh, S.; Nury, T.; Yammine, A.; Zaibi, M.; Mihoubi, W. 7-Ketocholesterol and 7β-hydroxycholesterol: In vitro and animal models used to characterize their activities and to identify molecules preventing their toxicity. Biochem. Pharmacol. 2020, 173, 113648.
- Doria, M.; Maugest, L.; Moreau, T.; Lizard, G.; Vejux, A. Contribution of cholesterol and oxysterols to the pathophysiology of Parkinson’s disease. Free Radic. Biol. Med. 2016, 101, 393–400.
- Jarvis, S.; Williamson, C.; Bevan, C.L. Liver X receptors and male (In) fertility. Int. J. Mol. Sci. 2019, 20, 5379.
- Ma, L.; Nelson, E.R. Oxysterols and nuclear receptors. Mol. Cell. Endocrinol. 2019, 484, 42–51.
- Bensinger, S.J.; Bradley, M.N.; Joseph, S.; Zelcer, N.; Janssen, E.; Hausner, M.A.; Shih, R.; Parks, J.S.; Edwards, P.A.; Jamieson, B.D.; et al. LXR Signaling Couples Sterol Metabolism to Proliferation in the Acquired Immune Response. Cell 2008, 134, 97–111.
- Sacchetti, P.; Sousa, K.M.; Hall, A.C.; Liste, I.; Steffensen, K.; Theofilopoulos, S.; Parish, C.; Hazenberg, C.; Richter, L.; Hovatta, O.; et al. Liver X Receptors and Oxysterols Promote Ventral Midbrain Neurogenesis In Vivo and in Human Embryonic Stem Cells. Cell Stem Cell 2009, 5, 409–419.
- Levy, D.; Ruiz, J.; Celestino, A.T.; Silva, S.F.; Ferreira, A.K.; Isaac, C.; Bydlowski, S.P. Short-term effects of 7-ketocholesterol on human adipose tissue mesenchymal stem cells in vitro. Biochem. Biophys. Res. Commun. 2014, 446, 720–725.
- Brahmi, F.; Vejux, A.; Sghaier, R.; Zarrouk, A.; Nury, T.; Meddeb, W.; Rezig, L.; Namsi, A.; Sassi, K.; Yammine, A.; et al. Prevention of 7-ketocholesterol-induced side effects by natural compounds. Crit. Rev. Food Sci. Nutr. 2018, 59, 3179–3198.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541.
- Danial, N.N.; Hockenbery, D.M. Chapter 18—Cell death. In Hematology, 7th ed.; Hoffman, R., Benz, E.J., Silberstein, L.E., Heslop, H.E., Weitz, J.I., Anastasi, J., Salama, M.E., Abutalib, S.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 186–196.
- Zarrouk, A.; Ben Salem, Y.; Hafsa, J.; Sghaier, R.; Charfeddine, B.; Limem, K.; Hammami, M.; Majdoub, H. 7β-hydroxycholesterol-induced cell death, oxidative stress, and fatty acid metabolism dysfunctions attenuated with sea urchin egg oil. Biochimie 2018, 153, 210–219.
- Paz, J.L.; Levy, D.; Oliveira, B.A.; Melo, T.; De Freitas, F.A.; Reichert, C.O.; Rodrigues, A.; Pereira, J.; Bydlowski, S.P. 7-Ketocholesterol Promotes Oxiapoptophagy in Bone Marrow Mesenchymal Stem Cell from Patients with Acute Myeloid Leukemia. Cells 2019, 8, 482.
- Sharma, B.; Agnihotri, N. Role of cholesterol homeostasis and its efflux pathways in cancer progression. J. Steroid Biochem. Mol. Biol. 2019, 191, 105377.
- Vejux, A.; Malvitte, L.; Lizard, G. Side effects of oxysterols: Cytotoxicity, oxidation, inflammation, and phospholipidosis. Braz. J. Med. Biol. Res. 2008, 41, 545–556.
- Green, D.R.; Llambi, F. Cell death signaling. Cold Spring Harbor Perspect. Biol. 2015, 7, a006080.
- Castelli, G.; Pelosi, E.; Testa, U. Emerging Therapies for Acute Myelogenus Leukemia Patients Targeting Apoptosis and Mitochondrial Metabolism. Cancers 2019, 11, 260.
- Schwartz, L.M.; Smith, S.W.; Jones, M.; Osborne, B.A. Do all programmed cell deaths occur via apoptosis? Proc. Natl. Acad. Sci. USA 1993, 90, 980–984.
- Esposti, M.D. Apoptosis: Who was first? Cell Death Differ. 1998, 5, 719.
- Wlodkowic, D.; Skommer, J.; Darzynkiewicz, Z. Cytometry of apoptosis. Historical perspective and new advances. Exp. Oncol. 2012, 34, 255–262.
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wideranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257.
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87.
- Dasgupta, A.; Nomura, M.; Shuck, R.; Yustein, J. Cancer’s Achilles’ heel: Apoptosis and necroptosis to the rescue. Int. J. Mol. Sci. 2017, 18, 23.
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516.
- Ashrafi, G.H.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2012, 20, 31–42.
- Corsetti, G.; Chen-Scarabelli, C.; Romano, C.; Pasini, E.; Dioguardi, F.S.; Onorati, F.; Knight, R.; Patel, H.; Saravolatz, L.; Faggian, G.; et al. Autophagy and Oncosis/Necroptosis Are Enhanced in Cardiomyocytes from Heart Failure Patients. Med. Sci. Monit. Basic Res. 2019, 25, 33–44.
- Kirkin, V. History of the selective autophagy research: How did it begin and where does it stand today? J. Mol. Biol. 2020, 432, 3–27.
- Jha, S.; Kitsis, R.N. Myocardial Basis for Heart Failure. Role of Cell Death. In Heart Failure; Elsevier Inc.: Philadelphia, PA, USA, 2011; pp. 85–102.
- Klionsky, D.J. Autophagy revisited: A conversation with Christian de Duve. Autophagy 2008, 4, 740–743.
- Pan, H.; Cai, N.; Li, M.; Liu, G.H.; Izpisua Belmonte, J.C. Autophagic control of cell ‘stemness’. EMBO Mol. Med. 2013, 5, 327–331.
- Gump, J.M.; Thorburn, A. Autophagy and apoptosis: What is the connection? Trends Cell Biol. 2011, 21, 387–392.
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036.
- Yun, C.W.; Lee, S.H. The roles of autophagy in cancer. Int. J. Mol. Sci. 2018, 19, 3466.