Human immunodeficiency virus type-1 (HIV-1) can undergo either a lytic pathway to cause productive systemic infection or enter a latent state in which the integrated provirus remains transcriptionally silent for decades.
The ability to latently infected T-cells enables HIV-1 to establish persistent infections in resting memory CD4+ T-lymphocytes which become reactivated following disruption or cessation of intensive drug therapy.
Maintenance of viral latency occurs through epigenetic and non-epigenetic mechanisms.
Epigenetic mechanisms of HIV latency regulation involve deacetylation and methylation of histone proteins within Nucleosome 1 (nuc -1) at the viral long terminal repeats (LTR) such that inhibition of histone deacetyltransferase and histone lysine methyltransferase activities, respectively, reactivates HIV from latency.
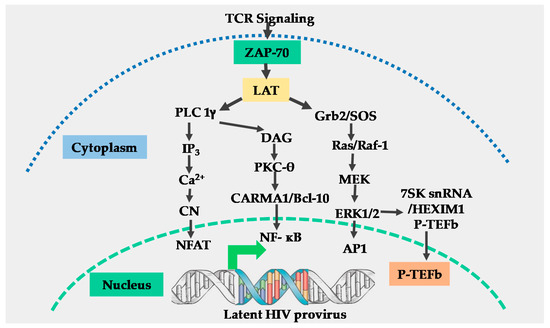
Non-epigenetic mechanisms involve nuclear restriction of critical cellular transcription factors such as Nuclear factor-kappa Beta (NF-kB) or Nuclear factor of activated T-cells (NFAT) which activate transcription from the viral LTR, limiting nuclear levels of viral transcription transactivator protein Tat and its cellular co-factor; positive transcription elongation factor b (P-TEFb) which together regulate HIV transcriptional elongation.
The T-cell receptor (TCR) activation efficiently induces NF-kB, NFAT, and activator protein 1 (AP-1) transcription factors through multiple signal pathways and how these factors efficiently regulate HIV LTR transcription through the non-epigenetic mechanism.
Elongation factor P-TEFb induced through an extracellular signal-regulated kinase (ERK) dependent mechanism regulates HIV transcriptional elongation before Tat is synthesized and the role of AP-1 in the modulation of HIV transcriptional elongation through functional synergy with NF-kB.
The TCR signaling induces critical posttranslational modifications of the Cyclin-dependent kinase 9 (CDK9) subunit of P-TEFb which enhances interactions between P-TEFb and viral Tat protein and the resultant enhancement of HIV transcriptional elongation.
- T-cell receptor signaling
- HIV,
- Transcription
1. Introduction
2. The T-Cell Receptor Signalosome
References
- Quinn, T.C. HIV epidemiology and the effects of antiviral therapy on long-term consequences. AIDS 2008, 22 (Suppl. 3), S7–S12. [Google Scholar] [CrossRef]
- Pau, A.K.; George, J.M. Antiretroviral therapy: Current drugs. Infect. Dis. Clin. N. Am. 2014, 28, 371–402. [Google Scholar] [CrossRef]
- Persaud, D.; Siberry, G.K.; Ahonkhai, A.; Kajdas, J.; Monie, D.; Hutton, N.; Watson, D.C.; Quinn, T.C.; Ray, S.C.; Siliciano, R.F. Continued production of drug-sensitive human immunodeficiency virus type 1 in children on combination antiretroviral therapy who have undetectable viral loads. J. Virol. 2004, 78, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Geeraert, L.; Kraus, G.; Pomerantz, R.J. Hide-and-seek: The challenge of viral persistence in HIV-1 infection. Annu. Rev. Med. 2008, 59, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Dornadula, G.; Zhang, H.; VanUitert, B.; Stern, J.; Livornese, L., Jr.; Ingerman, M.J.; Witek, J.; Kedanis, R.J.; Natkin, J.; DeSimone, J.; et al. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA 1999, 282, 1627–1632. [Google Scholar] [CrossRef] [PubMed]
- Sahu, G.K. Potential implication of residual viremia in patients on effective antiretroviral therapy. AIDS Res. Hum. Retrovir. 2015, 31, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Hokello, J.; Sharma, A.L.; Dimri, M.; Tyagi, M. Insights into the HIV Latency and the Role of Cytokines. Pathogens 2019, 8, 137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ramratnam, B.; Tenner-Racz, K.; He, Y.; Vesanen, M.; Lewin, S.; Talal, A.; Racz, P.; Perelson, A.S.; Korber, B.T.; et al. Quantifying residual HIV-1 replication in patients receiving combination antiretroviral therapy. N. Engl. J. Med. 1999, 340, 1605–1613. [Google Scholar] [CrossRef]
- Vanhamel, J.; Bruggemans, A.; Debyser, Z. Establishment of latent HIV-1 reservoirs: What do we really know? J. Virus Erad. 2019, 5, 3–9. [Google Scholar] [CrossRef]
- Tyagi, M.; Pearson, R.J.; Karn, J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J. Virol. 2010, 84, 6425–6437. [Google Scholar] [CrossRef]
- Tyagi, M.; Bukrinsky, M. Human immunodeficiency virus (HIV) latency: The major hurdle in HIV eradication. Mol. Med. 2012, 18, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Ruelas, D.S.; Greene, W.C. An integrated overview of HIV-1 latency. Cell 2013, 155, 519–529. [Google Scholar] [CrossRef]
- Tyagi, M.; Iordanskiy, S.; Ammosova, T.; Kumari, N.; Smith, K.; Breuer, D.; Ilatovskiy, A.V.; Kont, Y.S.; Ivanov, A.; Uren, A.; et al. Reactivation of latent HIV-1 provirus via targeting protein phosphatase-1. Retrovirology 2015, 12, 63. [Google Scholar] [CrossRef] [PubMed]
- Kabouridis, P.S. Lipid rafts in T cell receptor signalling. Mol. Membr. Biol. 2006, 23, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Tseng, S.Y.; Dustin, M.L. T-cell activation: A multidimensional signaling network. Curr. Opin. Cell Biol. 2002, 14, 575–580. [Google Scholar] [CrossRef]
- Harder, T. Lipid raft domains and protein networks in T-cell receptor signal transduction. Curr. Opin. Immunol. 2004, 16, 353–359. [Google Scholar] [CrossRef]
- Zhang, J.T.; Han, E.; Liu, Y. Role of the ribosome in sequence-specific regulation of membrane targeting and translocation of P-glycoprotein signal-anchor transmembrane segments. J. Cell Sci. 2000, 113 Pt 14, 2545–2555. [Google Scholar]
- Zhang, W.; Samelson, L.E. The role of membrane-associated adaptors in T cell receptor signalling. Semin. Immunol. 2000, 12, 35–41. [Google Scholar] [CrossRef]
- Kim, Y.K.; Mbonye, U.; Hokello, J.; Karn, J. T-cell receptor signaling enhances transcriptional elongation from latent HIV proviruses by activating P-TEFb through an ERK-dependent pathway. J. Mol. Biol. 2011, 410, 896–916. [Google Scholar] [CrossRef]
- Kane, L.P.; Shapiro, V.S.; Stokoe, D.; Weiss, A. Induction of NF-kappaB by the Akt/PKB kinase. Curr. Biol. CB 1999, 9, 601–604. [Google Scholar] [CrossRef]
- Narayan, P.; Holt, B.; Tosti, R.; Kane, L.P. CARMA1 is required for Akt-mediated NF-kappaB activation in T cells. Mol. Cell. Biol. 2006, 26, 2327–2336. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Abe, R. Signal Transduction Via Co-stimulatory and Co-inhibitory Receptors. Adv. Exp. Med. Biol. 2019, 1189, 85–133. [Google Scholar] [CrossRef] [PubMed]
- Harhaj, E.W.; Maggirwar, S.B.; Good, L.; Sun, S.C. CD28 mediates a potent costimulatory signal for rapid degradation of IkappaBbeta which is associated with accelerated activation of various NF-kappaB/Rel heterodimers. Mol. Cell. Biol. 1996, 16, 6736–6743. [Google Scholar] [CrossRef]
- Barat, C.; Tremblay, M.J. Engagement of CD43 enhances human immunodeficiency virus type 1 transcriptional activity and virus production that is induced upon TCR/CD3 stimulation. J. Biol. Chem. 2002, 277, 28714–28724. [Google Scholar] [CrossRef]
- Tardif, M.R.; Tremblay, M.J. Tetraspanin CD81 provides a costimulatory signal resulting in increased human immunodeficiency virus type 1 gene expression in primary CD4+ T lymphocytes through NF-kappaB, NFAT, and AP-1 transduction pathways. J. Virol. 2005, 79, 4316–4328. [Google Scholar] [CrossRef]
- Saito, T. Molecular Dynamics of Co-signal Molecules in T-Cell Activation. Adv. Exp. Med. Biol. 2019, 1189, 135–152. [Google Scholar] [CrossRef]
- Ormonde, J.V.S.; Li, Z.; Stegen, C.; Madrenas, J. TAOK3 Regulates Canonical TCR Signaling by Preventing Early SHP-1-Mediated Inactivation of LCK. J. Immunol. 2018, 201, 3431–3442. [Google Scholar] [CrossRef]
- Fenard, D.; Yonemoto, W.; de Noronha, C.; Cavrois, M.; Williams, S.A.; Greene, W.C. Nef is physically recruited into the immunological synapse and potentiates T cell activation early after TCR engagement. J. Immunol. 2005, 175, 6050–6057. [Google Scholar] [CrossRef]
- Neri, F.; Giolo, G.; Potesta, M.; Petrini, S.; Doria, M. The HIV-1 Nef protein has a dual role in T cell receptor signaling in infected CD4+ T lymphocytes. Virology 2011, 410, 316–326. [Google Scholar] [CrossRef]
- Fortin, J.F.; Barat, C.; Beausejour, Y.; Barbeau, B.; Tremblay, M.J. Hyper-responsiveness to stimulation of human immunodeficiency virus-infected CD4+ T cells requires Nef and Tat virus gene products and results from higher NFAT, NF-kappaB, and AP-1 induction. J. Biol. Chem. 2004, 279, 39520–39531. [Google Scholar] [CrossRef] [PubMed]
This entry is adapted from the peer-reviewed paper 10.3390/v12080868