2. AHR and Atopic Dermatitis
AD is a common and heterogenous eczematous skin disorder characterized by Th2-deviated skin inflammation, barrier disruption, and chronic pruritus [
17,
82,
83]. Frequent relapse with intense pruritus deteriorates quality of life and decreases treatment satisfaction of the afflicted patients [
84,
85,
86,
87,
88]. The lifetime incidence of AD is as high as 20% in the general population [
89]. Skin barrier dysfunction is associated with the reduced production of terminal differentiation molecules such as filaggrin [
15,
51]. Abnormal skin barrier integrity also causes an increased colonization of microbes such as
Staphylococcus aureus, which further exacerbate Th2-deviated skin inflammation [
90,
91]. In addition, some autoimmune diseases are comorbid with AD [
92].
Investigation on
AHR gene polymorphism reveals that
AHR rs10249788 and rs2066853 polymorphisms are found in patients with AD, psoriasis, and healthy controls, but no significant differences were detected in genotype or allele frequencies between the three groups [
93]. However, the
AHR rs2066853 (AG + AA) or rs10249788 (CT + TT) genotypes are a risk factor for severe dry skin phenotype and the combined rs10249788 (CT + TT) and rs2066853 (AG + AA) genotypes lead to a higher risk for severe dry skin in Chinese patients with AD [
93]. rs10249788 exists in the AHR promoter region where nuclear factor 1C (NF1C) binds and suppresses the transcription and protein expression of AHR [
94]. Notably, NF1C prefers to associate with the C allele compared to the T allele at rs10249788. Thus, subjects with the rs10249788 (CC) allele express less AHR than those with the rs10249788 (TT) allele [
94]. In fact, AHR mRNA levels for the TT genotype are 1.7-fold higher than those for the CC genotype [
95]. No significant differences were obtained in AHR production between the CC and CT genotypes [
95]. In parallel with increased levels of AHR, cells with the TT genotype express significantly higher levels of CYP1A1, IL-24, and IL-1β [
95]. It is intriguing that IL-24 downregulates the filaggrin expression via STAT3 activation [
67].
Immunohistological and real time PCR studies for AHR have been reported in AD [
96,
97]. Hong et al. showed an increased expression of both AHR and ARNT without CYP1A1 induction in the lesioned skin of AD compared with normal control skin [
96]. Alternatively, Kim et al. demonstrated an increased expression of ARNT and CYP1A1 but not AHR in the lesional skin of AD [
97]. As the Th2-deviated milieu potently reduces filaggrin and other barrier-related molecules, the upregulation of AHR/ARNT may be compensatory to attenuate the Th2-mediated filaggrin reduction. A recent study by Yu et al. demonstrated the possibility that the Th2-deviated milieu decreases the production of endogenous AHR ligand such as indole-3-aldehyde by commensal skin microbiota [
98]. These findings collectively suggest that most AHR likely lack physiological ligands in the Th2-prone milieu in AD. Therefore, rapid-metabolizing AHR ligands, such as FICZ and indole-3-aldehyde, appropriately activate the AHR/ARNT/FLG axis and may be beneficial in treating AD [
58,
98]. However, vigorous and long-lasting activation of the AHR/ARNT/FLG axis by slow-metabolizing dioxins and environmental pollutants may exacerbate barrier dysfunction and aggravate AD [
96,
99].
Although the pathogenic implication of AHR and its gene polymorphism in AD remain elusive, recent clinical trials using topical AHR ligand tapinarof have reported its efficacy for AD [
100,
101,
102]. Tapinarof (5-[(E)-2-phenylethenyl]-2-[propan-2-yl] benzene-1, 3-diol, WBI-1001, GSK2894512 or bentivimod) is a naturally derived (but is now a fully synthetic) hydroxylated stilbene produced by bacterial symbionts of entomopathogenic nematodes [
100,
101,
102,
103]. Tapinarof is a high affinity AHR ligand with antioxidative activity via NRF2 activation and a ROS-scavenging structure [
102] (
Figure 1). Tapinarof has gained increased attention because its topical application is efficacious for patients with AD in clinical trials [
21,
100,
104]. Tapinarof activates the AHR/CYP1A1 axis and augments the expression of filaggrin and involucrin [
102]. Even in barrier-disrupted AD patients, systemic absorption of topical tapinarof is limited and likely decreases during the treatment course in parallel with treatment success that restores the barrier dysfunction [
104]. In general, topical tapinarof is tolerable but frequent adverse events include headaches and folliculitis [
104].
In an early clinical trial, patients with AD affecting 3–20% of their body surface area (BSA) and with an Investigator’s Global Assessment (IGA; 0: clear, 1: almost clear, 2: mild, 3: moderate, 4: severe, 5: very severe) of 2–4 were randomized (1:1:1) to receive a placebo (
n = 51), topical tapinarof 0.5% (
n = 50) or 1% (
n = 47) in a cream formulation applied twice daily for six weeks [
100]. There was a decrease of 1.3 (43%;
p < 0.001; 95% confidence interval (CI) −1.2 to −0.5) and 1.8 (56.3%;
p < 0.001; 95% CI −1.6 to −0.9) in IGA at day 42 in the topical tapinarof 0.5% and 1% groups, respectively, compared with a decrease of 0.5 (14.7%) in the placebo group. At day 42, improvement in Eczema Area and Severity Index (EASI) score was 68.9% (
p < 0.001) and 76.3% (
p < 0.001) for tapinarof 0.5% and 1%, respectively, compared with 23.3% for placebo. Improvement in pruritus severity score at day 42 was 29.8% (
p < 0.001) and 66.9% (
p < 0.001) for tapinarof 0.5% and 1%, respectively, compared with 9.5% for placebo [
100]. Adverse events included headaches (placebo: 0%; 0.5% tapinarof: 8%; 1% tapinarof: 14%), migraines (placebo: 0%; 0.5% tapinarof: 4%; 1% tapinarof: 3%), folliculitis (placebo: 0%; 0.5% tapinarof: 6%; 1% tapinarof: 8%), and contact dermatitis (placebo: 0%; 0.5% tapinarof: 3%: 1% tapinarof: 5%) [
100].
A phase II, double-blind, vehicle-controlled, randomized, six-arm trial (1:1:1:1:1:1) in patients aged 12 to 65 years, with BSA involvement of at least 5% to 35% and an IGA score of 3 or higher (moderate to severe) at baseline was performed. Primary end points included an IGA score of clear or almost clear (0 or 1) and a minimum two-grade improvement (treatment success) at week 12 [
21]. The rates of treatment success with topical tapinarof cream at week 12 were 53% (1% twice daily,
n = 40), 46% (1% once daily,
n = 41), 37% (0.5% twice daily,
n = 43), 34% (0.5% once daily,
n = 41), 24% (vehicle twice daily,
n = 42), and 28% (vehicle once daily,
n = 40). The rate with tapinarof 1% twice daily (53%) was statistically significantly higher than the rate with vehicle twice daily (24%). Notably, treatment success was maintained for four weeks after the end of tapinarof treatment. The proportion of patients achieving EASI75 (75% or greater improvement in EASI) score reduction at week 12 was significantly higher in the groups treated with 1% tapinarof (60% and 51% for twice daily and once daily, respectively) than with vehicle (26% and 25% in the groups receiving vehicle twice daily and once daily, respectively) [
21]. Headaches (e.g., 10% (1% twice daily), 2% (0.5% twice daily), and 0% (0.5% twice daily)) and folliculitis (e.g., 10% (1% twice daily), 7% (0.5% twice daily), and 0% (0.5% twice daily)) were again frequent adverse events [
21].
In a murine dermatitis model, topically applied FICZ activated AHR and significantly reduced the dermatitis score and histological inflammation with a decrease of
Il22 gene expression in chronic mite antigen-induced dermatitis [
58]. In addition, topical FICZ restored the dermatitis-induced filaggrin downregulation [
58]. CCL17 and CCL22 are crucial chemokines to recruit Th2 cells [
68]. IL-4/IL-13 stimulates dendritic cells to produce CCL17 and CCL22 via STAT6 activation and contributes to the recruitment of Th2 cells in the lesional skin of AD [
68]. Soybean tar Glyteer inhibits the IL-4/IL-13-mediated STAT6 activation and subsequent production of CCL17 and CCL22 in dendritic cells [
68]. In addition, pruritogenic Th2 cytokine IL-31 synergistically upregulates the IL-4/IL-13-mediated CCL17 and CCL22 production in dendritic cells because IL-4/IL-13 increase IL-31 receptor A (IL31RA) expression [
105]. Glyteer again attenuates the IL-4/IL-13-mediated IL31RA upregulation and subsequent CCL17 and CCL22 production by inhibiting STAT6 activation [
105]. It is known that coal tar inhibits STAT6 activation via the NRF2-antioxidative pathway [
15]. Ligation of AHR by FICZ also reduces the expression of type 1 IgE Fc receptor in Langerhans cells [
106].
Although antioxidative AHR ligands are therapeutic for dermatitis, exaggerated activation of AHR by genetic manipulation in transgenic mice or by dioxin treatment induces itchy dermatitis most likely due to an abnormally accelerated keratinization process, epidermal acanthosis, elongation of nerve fibers, and production of pruritogenic artemin [
47,
99,
107]. Therefore, extreme activation of AHR is deleterious for skin. In parallel, ovalbumin-induced delayed hypersensitivity is enhanced by topical benzopyrene with upregulation of IL-5, IL-13, and IL-17 expression in lymph node cells [
96].
Since FICZ is an endogenous UVB photoproduct [
8], the barrier-protecting effects of FICZ may explain, at least in part, why UVB phototherapy is efficacious for the treatment of AD and psoriasis [
108,
109].
3. AHR and Psoriasis
Psoriasis is an (auto)immune-mediated disease that manifests as widespread desquamative erythema [
110,
111]. Males are twice as likely to be affected than females [
112,
113]. The cosmetic disfigurement associated with psoriasis profoundly impairs the patients’ quality of life, treatment satisfaction and adherence, and socioeconomic stability [
114,
115]. The autoimmune nature of psoriasis is exemplified by its high comorbidity with psoriatic arthritis [
110,
116,
117,
118] and other autoimmune diseases including autoimmune bullous diseases [
119,
120,
121,
122,
123,
124]. Psoriasis is also comorbid with cardiovascular diseases, metabolic diseases, and renal disorders, which represent a condition called inflammatory skin march [
111,
125,
126,
127,
128,
129]. The excellent therapeutic efficacy of anti-TNF-α/IL-23/IL-17A biologics for psoriasis point to the central role of the TNF-α/IL-23/IL-17A axis in its pathogenesis [
18,
19,
130,
131,
132,
133,
134] Additionally, genetic and environmental factors are known to be involved in its pathogenesis [
135,
136].
As AHR predominantly regulates the immune balance of Th17/22 and Treg cells [
28,
29,
69,
70], AHR is expected to play a significant role in psoriasis [
102]. In an imiquimod-induced psoriasis model,
AhR deficiency exacerbates skin inflammation with upregulated gene expression of
Il22,
Il17a, and
Il23 [
137]. The intensity of delayed type-hypersensitivity is also enhanced in
Ahr-deficient mice [
137]. However, further experiments demonstrated that
Ahr-deficiency in nonhematopoietic cells, including keratinocytes, but not in hematopoietic cells, was likely responsible for the exacerbation of inflammation [
137]. Notably, intraperitoneal injection of FICZ ameliorated the imiquimod-induced psoriasis-like inflammation. Tapinarof and FICZ also reduced the imiquimod-induced psoriasiform skin inflammation by inhibiting
Il17a,
Il17f,
Il19,
Il22,
Il23a, and
Il1b gene expression [
102]. The therapeutic action of tapinarof and FICZ was AHR-dependent because it was not observed in
Ahr-deficient mice [
102]. In an ex vivo activation assay of skin-resident immunocompetent cells using normal human skin, tapinarof inhibited the expression of
IL17A message approximately 50% but increased the
IL22 expression [
102,
138] (
Figure 1). In mice, IL-22 is produced from Th17, γδT, ILC3, and CD4
−CD8
−TCRβ
+ cells [
139]. AHR was required for IL-22 production by Th17, but not by the three other cell types, in the imiquimod-treated ears [
139]. Although imiquimod-induced skin inflammation is popular as a psoriasis model, attention should be paid because imiquimod is degraded by CYP1A1 so the efficacy of AHR agonists may partly rely on this effect in the imiquimod model [
140].
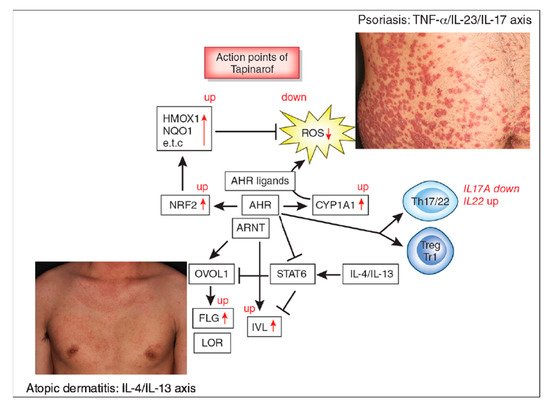
Figure 1. Aryl hydrocarbon receptor (AHR) signal and action points of tapinarof (red words and arrows). AHR is a promiscuous chemical sensor that is activated by various oxidative and antioxidative ligands. Once activated, cytoplasmic AHR translocates into the nucleus where it heterodimerizes with an AHR-nuclear translocator (ARNT) and then induces the transcription of AHR-responsive genes such as cytochrome P450 1A1 (CYP1A1). CYP1A1 degrades AHR ligands. Some ligands such as dioxins are chemically stable and long-lived. Therefore, CYP1A1 generates high amounts of reactive oxygen species (ROS) after sustained efforts to degrade them. Some antioxidative AHR ligands activate nuclear factor-erythroid 2-related factor-2 (NRF2) transcription factor, which upregulates gene expression of various antioxidative enzymes, such as heme oxygenase 1 (HMOX1), NAD(P)H dehydrogenase, and quinone 1 (NQO1), and these antioxidative enzymes neutralize ROS. AHR/ARNT signaling also activates OVO-like 1 (OVOL1) transcription factor and upregulates the expression of filaggrin (FLG) and loricrin (LOR). AHR upregulates the expression of involucrin (IVL) in an OVOL1-independent manner. Therefore, AHR/ARNT signaling accelerates epidermal terminal differentiation and enhances the repair of barrier disruption. Interleukin (IL)-4 and IL-13 activate signal transducer and activator of transcription 6 (STAT6) and inhibit the OVOL1/FLG, OVOL1/LOR, and AHR/IVL axes. However, suitable AHR activation can inhibit the IL-4/IL-13-mediated STAT6 activation and restore the expression of FLG, LOR, and IVL. Regarding immune response, AHR signaling affects T-helper (Th17) differentiation and is essential for IL-22 production. AHR ligation (especially by high concentrations of ligands) induces the differentiation of regulatory cell populations, Treg and Tr1 cells. Tapinarof is an antioxidative AHR ligand and upregulates CYP1A1 expression. Topical tapinarof is efficacious in psoriasis and atopic dermatitis. Current studies demonstrate that tapinarof activates NRF2/antioxidative signaling and reduces oxidative stress. Tapinarof also upregulates FLG and IVL expression. Tapinarof downregulates IL-17A production and increases IL-22 production.
Immunohistological and real time PCR studies have demonstrated that the expression of AHR and ARNT is upregulated in the lesional skin of psoriasis, whereas CYP1A1 expression was significantly decreased compared to normal controls [
97]. In contrast, serum levels of both AHR and CYP1A1 are elevated in patients with psoriasis compared to normal controls [
141]. Further studies are warranted to investigate these controversial data.