Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Hydrogen sulfide (H2S) is a colorless, corrosive gas with a characteristic rotten egg smell, known for over 300 years as an environmental toxin [1]. High H2S concentrations cause damage in many organs, including the brain, kidneys, and lungs.
- hydrogen sulfide
- H2S
- cancer
- cystathionine β-synthase
- cystathionine γ-lyase
- 3-mercaptopyruvate sulfurtransferase
1. H2S Chemistry, Synthesis, and Molecular Mechanisms of Action
1.1. H2S Chemistry
The biochemistry of H2S is ancient and was likely essential in the development of life along with its antecedent, pre-biotic chemistry [17]. H2S is a weak diprotic acid, with first and second pKa values of 6.76 and 19 at 37 °C. At the physiologic pH H2S is ~70–80% HS−, ~20% H2S, with low concentrations of S2− [18]. H2S readily passes through biological membranes allowing for efficient paracrine signaling, while HS− has a high nucleophilicity, conferring chemical reactivity [19,20]. Biological membranes are not significant barriers to H2S diffusion, possibly due to the rapid equilibrium between HS− and H2S [19]. The minus two oxidation state of the sulfur in H2S is the lowest possible sulfur oxidation state, making H2S an obligatory reductant [7]. H2S-oxidant interactions have been examined in many studies, where H2S is reported to have antioxidant effects [21,22,23,24]. Interestingly, H2S and H2O2 react too slowly to exert significant biologic effects [25,26]. Moreover, cellular H2S concentrations are low at 10–30 nM, compared to other thiol-based antioxidants, such as glutathione at 1–10 mM. Thus, under biologic conditions it is unlikely that free H2S has a quantitatively significant antioxidant role [27,28]. Most likely, a significant portion of H2S-mediated antioxidant activity is due to its induction of other cellular antioxidant defense systems, such as increasing cystine uptake, and cellular glutathione (GSH) and cysteine concentrations [7,21]. Additionally, free H2S exists in a thermodynamic equilibrium with many other sulfur species, including persulfides, polysulfides, and numerous sulfide metabolites, many of which are antioxidants [7]. In most tissues the H2S half-life is short, lasting only a few minutes [7,29].
1.2. H2S Synthesis
H2S is chiefly synthesized by three systems: (1) cellular enzymes, (2) nonenzymatic synthesis, and (3) production by the microbiome. Enzymatic H2S synthesis is based on the reverse transsulfuration pathway, where dietary methionine is converted into homocysteine. Cystathionine β-synthase (CBS) then catalyzes the condensation of homocysteine with cysteine, yielding cystathionine and H2S [30]. Cystathionine γ-lyase (CSE) catalyzes the conversion of cystathionine into α-ketobutyrate, cysteine, and NH3 [30]. CBS and CSE are both pyridoxal 5′-phosphate-dependent enzymes and can catalyze other substrates, depending on the specific substrate concentrations [7,31]. For example, CSE also catalyzes the conversion of L-cysteine to thiocysteine, pyruvate, and NH3. Thiocysteine in turn can non-enzymatically break down into pyruvate and H2S. H2S is also enzymatically synthesized by cysteine aminotransferase which converts cysteine to mercaptopyruvate, which 3-mercaptopyruvate sulfurtransferase (3-MST) in turn converts into H2S and pyruvate [7,31].
CBS, CSE, and 3-MST show distinct tissue and subcellular distributions and exert different contributions to specific cell/tissue H2S pools. 3-MST is primarily mitochondrial and synthesizes roughly 90% of brain H2S [6,32]. CBS exists within many tissues but is predominantly seen in the central nervous system and liver, while CSE is predominantly located in the vasculature [30,31]. Intracellular H2S exists as free H2S, acid-labile sulfide, and bound sulfane sulfur [29]. Free cellular H2S represents less than 1% of the potentially available sulfide, indicating that the endogenous sulfide pool likely has significant buffering capacity [7].
CBS, CSE, and 3-MST also synthesize polysulfides. In the brain, 3-MST synthesizes H2S3, which is known to alter the activities of proteins, such as KEAP1, PTEN, and GAPDH [33,34]. CSE overexpressing human A549 carcinoma cells have high polysulfide levels, while CSE knockout cells show significantly lower levels, and recently CSE-generated polysulfides have been shown to play a role in ovarian cancer [35,36]. Additionally, enzymatic kinetic analyses have shown that polysulfides are likely products of CBS and CSE activities [34]. Last, the enzyme cysteinyl tRNA synthetase produces protein hydropersulfides, which are involved in the regulation of mitochondrial biogenesis and bioenergetics, and many other cellular functions [37].
Nonenzymatic H2S synthesis also accounts for a portion of H2S synthesis and occurs by an iron and vitamin B6-mediated catalysis of thiol and thiol-containing compounds, such as cysteine [38]. Last, the mammalian microbiome regulates the bioavailability and metabolism of systemic H2S. For example, compared to conventionally housed mice, germ-free mice exhibit significantly lower plasma and gastrointestinal free H2S, and 50–80% lower plasma, adipose, and lung tissue bound sulfane sulfur. Tissue CSE activity was also reduced in many of the organs in germ-free mice, while tissue cysteine levels were elevated [39].
1.3. Molecular Mechanisms of H2S Activity
Based on its chemistry, the molecular mechanisms of H2S reactivity have been placed into three categories: (1) chemical interfacing/scavenging with reactive oxygen and nitrogen species, (2) chemical modification of protein cysteines to persulfides, and (3) binding to and/or redox reactions with metal centers [7,20]. Additionally, H2S may chemically reduce protein disulfides. The biochemical significance of this mechanism is currently unknown, and if it occurs, it is likely highly specific to the chemical milieu of the protein disulfide [40].
2. H2S and Cancer
Recently, a complex and intricate role for H2S has been identified in cancer. Below we discuss the role of H2S in several common human malignancies and discuss the functions of H2S in the initiation and promotion of cancer.
3.1. H2S and Colorectal Cancer
Colon cancer is the second most lethal and third most common cancer worldwide [41]. The role of H2S in cancer was demonstrated in 2013 when increased cytoplasmic and mitochondrial CBS expression was detected in seven colorectal carcinoma resections, compared to benign adjacent colonic tissues. CSE and 3-MST levels were unchanged. Several malignant colon cancer-derived cell lines similarly showed high CBS expression. Tumor tissue lysates incubated with L-cysteine and L-homocysteine also produced 6-fold more H2S than lysates from benign tissue. CBS knockdown or pharmacologic inhibition by aminooxyacetic acid (AOAA) reduced cell proliferation and H2S synthesis in the HCT116 colon cancer cell line, but not in the slower-growing nonmalignant NCM356 cell line. CSE knockdown or pharmacologic inhibition by DL-propargyl glycine (PAG) affected neither HCT116 cell proliferation nor H2S synthesis. AOAA also suppressed the migration and invasion of HCT116 cells, while NaHS, an H2S donor, enhanced these events [42]. Low to moderate concentrations (0.1–1 mM) of the CBS allosteric activator S-adenosyl-L-methionine also increased HCT116 cell proliferation, bioenergetics, and H2S synthesis for up to twelve hours, an event not seen with CBS knockdown [42,43].
CBS knockdown or AOAA inhibition also attenuated mitochondrial function, suppressing basal respiration, ATP production, and the spare respiratory capacity. CBS knockdown similarly reduced glycolysis, in part through reduced glyceraldehyde 3-phosphate dehydrogenase (GAPDH) activity. Cystathionine failed to stimulate cell proliferation or cellular bioenergetics, indicating that H2S and not cystathionine affected these cell behaviors. CBS, but not CSE knockdown, also significantly reduced the growth and volume of HCT116 cell xenografts in Nu/nu Balb/C female mice, with a concomitant reduction in CD31-immunopositive blood vessel density, the prevalence of larger blood vessels, and blood vessel branching. Additionally, the direct injection of AOAA into the xenograft tumor parenchyma reduced peritumor blood flow, while administration of the CBS substrate L-cysteine increased this flow [42,43].
Further studies revealed that premalignant colonic tubular adenomas (low-grade epithelial dysplasia) exhibited increased CBS expression, indicating that increased CBS expression is an early event in colon cancer [44]. Lentiviral vector-mediated CBS expression in the nonmalignant NCM356 colon cell line increased H2S synthesis, reverse transsulfuration pathway flux, cell proliferation, migration, invasion, mitochondrial bioenergetics, and glycolysis. CBS expressing NCM356 cells also formed larger tumors in athymic nude mice compared to vector only bearing cells. Whole transcriptome RNA sequencing was employed to compare the CBS expressing NCM356 cells to those carrying the vector alone. CBS expression caused a metabolic shift towards anabolic metabolism, increasing the gene expression patterns related to glycolysis, hypoxic responses, extracellular matrix formation, cell adhesion molecule production, and epithelial to mesenchymal transition (EMT), resulting in a gene expression pattern that overlapped with an oncogenic phenotype [44].
These studies were extended when HCT116 cells were treated with 300 μM of the 3-MST inhibitor 2-[(4-hydroxy-6-methylpyrimidin-2-yl)sulfanyl]-1-(naphthalen-1-yl)ethan-1-one (HMPSNE). HMPSNE suppressed 3-MST, CBS, the H2S-degrading enzymes ethylmalonic encephalopathy 1 protein, and rhodanese expression, suggesting that 3-MST-derived (but not CBS/CSE-derived) H2S exerts transcriptional regulatory actions in these cells [45]. HMPSNE treatment also increased mRNA and protein expression of the epithelial markers E-cadherin and tight junction protein ZO-1. Concomitantly, mRNAs for the mesenchymal markers fibronectin, Wnt3, β-catenin, and cadherin-associated protein β1, the transcription factors Snail1, Twist1, Sp1, and Sp3, and β-catenin, and both the ATP citrate lyase (ACLY) protein and mRNA expression were suppressed. ACLY is a key enzyme in acetyl-CoA synthesis associated with Wnt signaling and is implicated in the colon cancer EMT [45]. ACLY enzymatic activity was not induced by H2S; however, H2S partially reversed its inactivation by reactive oxygen species (ROS). Moreover, the slow H2S donor GYY4137 activated the ACLY gene promotor through the induction of the Sp3 transcription factor. Lastly, HMPSNE treatment also attenuated cell migration, consistent with EMT inhibition [46]. Taken together, the above studies indicate that H2S promotes the progression of colon cancer from preneoplastic low-grade dysplastic lesions to metastatic, dedifferentiated colon cancer.
The inhibition of H2S synthesis increases the sensitivity colon cancer cells to chemotherapeutic agents, while chemotherapy resistance is associated with increased H2S synthesis. For example, the treatment of the colon cancer cell lines HT-29 and DLD-1 with AOAA increased their sensitivity to 5-fluorouracil (5-FU), lowering cell colony formation, Bcl-2 expression, and viability, while increasing cell S-phase accumulation, apoptosis, cytochrome C release, and Bax expression. In a murine tumor xenograft model, DLD-1 cells stably over-expressing luciferase were injected into mice, allowing the bioluminescent noninvasive imaging of the cells. AOAA and 5-FU treatments in this model both lowered tumor growth, while the two combined synergistically suppressed tumor growth. AOAA also suppressed the target of 5-FU, thymidylate synthase. Further analysis revealed that CBS-synthesized H2S suppressed the colon cancer tumor suppressor miR-215-5p, increasing both thymidylate synthase and epiregulin (an EGFR signaling activator) [47]. AOAA treatment also increased the sensitivity of the colon cancer cell line oxaliplatin, significantly reducing the IC50 value, and GSH, Bcl-2, total caspase-9, and total caspase-3 expression, while increasing oxaliplatin-induced apoptosis, ROS levels, cleaved caspase-9, cleaved PARP, Bax, and p53 expression. In murine xenograft studies, AOAA and oxaliplatin reduced colon cancer cell growth, while the combination significantly reduced tumor volume compared to either agent alone [48].
In an interesting study, HCT116 cells were cultured for six months with increasing 5-FU concentrations in media maintained at 30 μM of 5-FU. When challenged with 3–100 μM of 5-FU, the parental cells exhibited lower viability and proliferation, while the 5-FU resistant cells showed no viability or proliferative changes. Western blot analyses revealed that CBS, 3-MST, and rhodanese increased by 49%, 63%, and 107% in the 5-FU resistant cells, with higher CBS and 3-MST enzymatic activities in these cells [49]. The 5-FU resistant cell line showed increased glutamate oxaloacetate transaminase 1 and cytochromes p450 CYP1A2 and CYP2A6 expression. The p450 inhibitor phenylpyrrole sensitized the 5-FU resistant cells to 5-FU treatment, while the sensitivity of the parental cell line 5-FU was not increased by this treatment. Lastly, the 5-FU resistant cells were resistant to AOAA treatment, showing less viability and proliferation suppression than the parental cell line when treated with AOAA and 5-FU. Taken together, the above data indicate that increased CBS and 3-MST protein levels and enzymatic activities play a role in chemotherapy resistance [49]. The roles of CBS, 3-MST, and H2S in colon cancer are summarized in Figure 1.
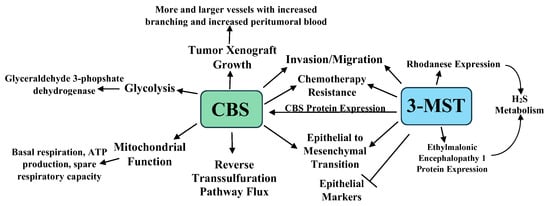
2.2. H2S and Ovarian Cancer
Ovarian cancer carries a 1.39% lifetime risk in women and due to its often-advanced stage at presentation, it is one of the most lethal gynecologic malignancies [50]. A tissue microarray analysis of 210 epithelial ovarian tumors revealed increased CBS expression compared to benign counterpart tissue. Moderate to strong CBS immunoreactivity was associated with a serous histology and a higher tumor grade, although significant expression was also seen in early-stage tumors. Additionally, of six ovarian tumor cell lines examined, four showed significant CBS protein and mRNA expression, while two showed high CSE expression compared to a benign epithelial ovarian cell line. CBS siRNA knockdown or AOAA treatment of the CBS-expressing ovarian cancer cells lines resulted in a significant suppression of cell proliferation (measured by 3H-thymine incorporation) and a lowered viability. CBS siRNA knockdown also resulted in a significant reduction in the total cellular GSH (GSH + GSSH) and increased RelA/p65 expression (a subunit of NF-κB), with concomitant increased ROS and p53 protein expression. Interestingly, culturing the cells in 5 mM Na2S resulted in a 20% increase in cell viability and culturing in 2 mM of GSH-containing media resulted in a complete recovery of cell viability, indicating a link between CBS-synthesized H2S, GSH levels, and cell viability. Lastly, siRNA CBS KD also increased the sensitivity of the ovarian cancer cell line to cisplatin, lowering the IC50 from 13.1 to 7.9 μM [51].
The A2780 ovarian cancer cell line showed both cytoplasmic and mitochondrial CBS expression. CBS knockdown in these cells significantly enhanced MitoSOX fluorescence, indicating increased superoxide. Additionally, citrate synthase activity was also decreased with increased AOAA concentrations, suggesting that CBS knockdown inhibited mitochondrial oxidative capacities. To confirm this, siRNA CBS knockdown and scrambled RNA (scRNA)-treated cell mitochondrial bioenergetics were analyzed [51]. CBS knockdown decreased both basal and maximally stimulated mitochondrial respiration. These changes were accompanied by an increase in the NAD/NADH and ADP/ATP ratios and ROS levels, and a decrease in total ATP [51]. Taken together, these data demonstrate a role of CBS in the maintenance and regulation of mitochondrial function in ovarian cancer [51].
To extend these observations, the mitochondrial fusion and fission apparatus proteins MFN1, MFN2, Drp1, OPA1, and Fis1 were examined in two ovarian cancer cell lines (CP20 and OV90) with scRNA or CBS siRNA knockdown [52]. Of the five proteins, only MFN2 expression was down-regulated by CBS knockdown. Interestingly, the Mitotracker Red CMXRos staining of these cells revealed that the scRNA cells had fused and elongated the mitochondrial morphology, while the siRNA CBS knockdown cells displayed a predominantly spherical mitochondrial morphology and more unbranched mitochondria with a shorter length. Additionally, CBS or MRN2 siRNA knockdown significantly lowered the mitochondrial oxygen consumption rate and the membrane potential, cell proliferation, ATP production, and spare respiratory capacity. The treatment of siRNA CBS knockdown cells with the slow H2S donor GYY4137 increased the mitochondrial oxygen consumption rate, suggesting that CBS-generated metabolites regulate mitochondrial function. No changes in the autophagy markers or PGC1α were detected in the CBS knockdown cells. Similarly, the mitochondrial genome number did not change with CBS knockdown. Based on this, it was concluded that CBS knockdown induced mitochondrial fragmentation, but not autophagy by MFN2 suppression. Additionally, CBS maintained mitochondrial health through promoting fusion, ATP synthesis, and MFN2 stabilization [52].
CBS silencing did not affect MRN1 or MRN2 mRNA expression. Immunoprecipitation analyses revealed that CBS silencing increased MFN2 ubiquitination, resulting in its destabilization and degradation. Support for this came from the observation that treatment with the proteasome inhibitor MG132 rescued MFN2 expression with CBS silencing. The JNK kinase is activated by oxidative stress, resulting in its phosphorylation of MFN2, followed by MFN2 ubiquitination. Treatment of CBS silenced cells with either the antioxidant mitoTEMPO or the JNK inhibitor SP600125 rescued MFN2 expression [52]. Thus, CBS maintains mitochondrial function by attenuating ROS levels, an event required to preserved MFN2 protein expression [52]. The clinical relevance of CBS and MFN2 expression in ovarian cancer was examined in the clinically annotated mRNA data from The Cancer Genome Atlas (Gene Expression Omnibus) databases. In the quantitative analysis for Kaplan–Meier overall survival curves, a high expression of CBS and MFN2 conferred a poor overall survival of patients, with CBS and MFN2 expression showing a significant association in the same patient cohort [52].
The effects of CBS expression on tumor growth were examined in a previously developed murine orthotopic model of ovarian cancer. Mice were interperitoneally injected with the ovarian A2780 tumor cell line and after one week the mice were injected with neutral liposomes containing either control siRNA or CBS siRNA twice weekly, with or without cisplatin [52]. After four weeks the mice were sacrificed, and the tumors were analyzed. The mice receiving the CBS siRNA had a 40% lower tumor weight, a 70% reduction in tumor nodules, and lower proliferation and blood vessel formation, as measured by Ki-67 and CD31 immunostaining, respectively [52]. Interestingly, with cisplatin, the siRNA treated mice showed a small and nonsignificant reduction in tumor weight, while the mice treated with CBS siRNA plus cisplatin exhibited a 90% reduction in tumor weight compared to the mice treated with siRNA only [51]. The roles of CBS and H2S in ovarian cancer are summarized in Figure 2.
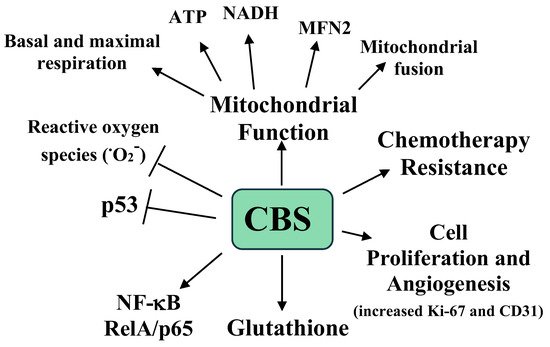
Figure 2. A summary of the roles of CBS and H2S in ovarian cancer growth, progression, metastasis, mitochondrial, function, and chemotherapy resistance. O2− is the superoxide radical [51].
This entry is adapted from the peer-reviewed paper 10.3390/pathophysiology28030028
This entry is offline, you can click here to edit this entry!