Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Protein kinases (PK) are signaling regulators involved in various cellular functions including metabolism, cell cycle regulation, survival, and differentiation. Tumor dependence of continuous proliferative signals mediated through protein kinases overexpression instigated increased strategies of kinase inhibition in the oncologic practice over the last couple decades.
- hematologic neoplasms
- targeted molecular therapy
- protein kinase inhibitors
- TKIs
1. Introduction
Cancer is a subset of noncommunicable diseases responsible for millions of deaths every year and is still seen as a major barrier to life expectancy increase worldwide [1][2]. Human tumors develop from healthy cellular populations after genetic deregulation at chromosomal or molecular levels that lead to increased cellular proliferation and overexpression of survival mechanisms. Complex interactions between heterogenic tumor cellular populations and tumor/human organism conceive biological advantages that ensure continuous growth in neoplastic clones, even in otherwise adverse scenarios [3][4].
Leukemias and lymphomas are a group of several hematologic and lymphoid tissue disorders that are characterized by accelerated expansion of clonal neoplastic populations of immunohematologic cell lines in the peripheral blood/bone marrow and lymph nodes of afflicted patients [5][6]. Non-Hodgkin’s lymphomas are among the 10 more incident cancers in the world, and, added together, leukemias afflicted more than 400,000 people every year, with diverse treatment efficacies for the different acute and chronic subtypes [2][6].
The ability of cancer cells to sustain continuous proliferative signals is a hallmark of cancer, and such ability is often dependent on the increased activities of growth factors and protein kinases (PK) [4]. Over the last couple decades, the advent of kinase inhibitor (KI) treatment in the oncologic practice represented a major step in targeted therapies development because it greatly enhanced the prognosis of many patients who used to depend on highly cytotoxic drug protocols for a chance of cancer treatment success [7][8].
1.1. Conventional Therapies and Resistance in Hematologic Neoplasms
The process of self-renewal of hematopoietic stem cells is essential to homeostasis maintenance. Once this process is disrupted and defects in cell differentiation occur, the malignant transformation and the appearance of hematologic neoplasms can be triggered [9]. Although these neoplasms may have unclear etiology, genetic alterations as chromosomal rearrangements, point mutations, aneuploidies, deletions, insertions, duplications, and/or amplifications are characteristics of hematologic neoplasms [10][11][12][13].
Leukemias were first described in 1827, and since then, treatment has changed dramatically. First, studies showed that some cases were treated on single-agent chemotherapy by using nitrogen mustard in Hodgkin’s disease and other lymphomas and chronic leukemias, as well as by administration of the folic acid antagonist aminopterin in acute leukemia patients [14][15][16][17]. The first leukemia treatment protocol was a combination of radiation, arsenic and thorium-X [15][16] and, even after some decades, arsenic continued as part of leukemia treatment. A study published later demonstrated the successful effect of arsenic in the treatment of promyelocytic leukemia, once 11 patients presented clinical remission after arsenic administration [18].
Then, research and development of new classes of antitumoral agents such as antimetabolites, antibiotics, and alkaloids with promising results in pre-clinical and clinical tests were inserted in leukemia treatment protocols, first as single agents and after in combination, with the perspective of prolonging or maintaining disease remission [19]. For example, the 1950s and 1960s introduced several new drugs to antileukemic chemotherapy as the use of cortisone, chlorambucil, mercaptopurine, cytoxan, vincristine, vinblastin, daunorubicin, and bleomycin to the treatment of leukemias and/or lymphomas [20][21][22][23][24][25][26], methotrexate and vincristine in childhood acute leukemia [27][28], L-asparaginase and daunorubicin in acute leukemia therapy [29][30]. In 1958, methotrexate was administered intrathecally to kill leukemic cells present in the central nervous system [31] and chlorambucil and cyclophosphamide were later used in the treatment of chronic granulocytic and lymphocytic leukemias, respectively [32][33].
The combined protocols were introduced at the end of the 1960s, with the administration of cyclophosphamide, mercaptopurine, methotrexate, and vincristine in acute lymphocytic leukemia in children [34], cyclophosphamide and vinca alkaloids in malignant lymphoma [35] and cytarabine (AraC) and daunorubicin in acute myeloid leukemia [36]. With the increase in the number of antileukemic drugs and treatment protocols available, and with the better understanding of hematologic neoplasm development, the survival rate improved expressively among pediatric leukemia patients, as well as in patients with Hodgkin’s disease and the non-Hodgkin’s lymphomas [15][37][38][39].
However, over time, cases of drug resistance to available chemotherapy were diagnosed among patients with relapsed leukemias. Then, protocols with more intensive chemotherapy and radiotherapy were evaluated, with extensively toxic effects on patients [19][40][41]. In the 1970s, Don Thomas presents bone marrow transplants as a complement to acute leukemia treatment, with promising results, but with several obstacles related to tissue typing, infection control, immunosuppression, transfusion support and the need to develop more specific drugs that escape from cellular resistance [15][42][43].
There are several factors related to cancer cell resistance to antitumoral compounds described in the literature. Those can be divided into intrinsic factors that are pre-existent to the treatment, and extrinsic ones that are acquired after starting chemotherapy (Figure 1). Together, those factors are responsible for treatment failure, cancer recurrences in the clinic and enhanced mortality rates among affected patients [44][45].
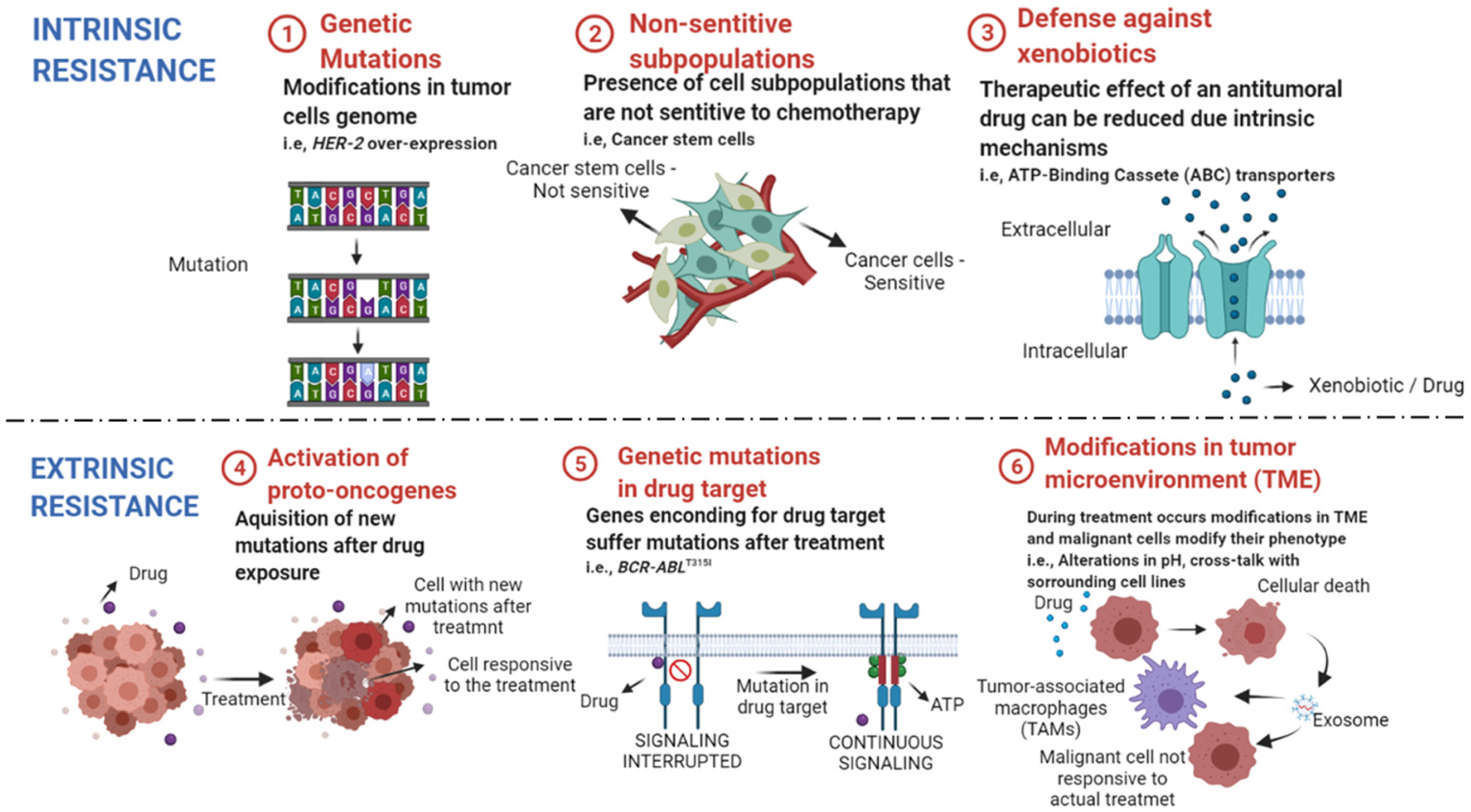
Figure 1. Intrinsic and extrinsic mechanisms of drug resistance in cancer. ① Genetic mutations can lead to alterations in the expression of genes related to cellular resistance and surveillance. ② Heterogenous tumors have subpopulations that may not respond to the available cytotoxic drugs, leading to cancer remission after treatment. ③ Some cellular transporters protect our cells from environmental toxins, as anticancer drugs, and reduce the concentration of their intracellular levels. ④ After treatment, new oncogenes can be activated, leading to enhanced proliferation rate of not responsive cells. ⑤ Mutations in genes that encode drug targets reduce drug efficacy in mutated cell lines. ⑥ Treatment can alter the tumor microenvironment (TME) and lead to cross-talk between sensitive cells and the surrounding cells. The exchange of resistance elements with tumor-associated macrophages (TAMs) and other tumoral cell lines lead to enhanced cell resistance to chemotherapy. Created with BioRender.com, accessed date: 20 August 2021.
Intrinsic resistance is characterized by resistance mechanisms inherent to the patient, once that is not triggered by the administration of chemotherapy drugs [46]. Factors such as (1) the existence of genetic mutations (mutations, gene amplifications, deletions, chromosomal rearrangements, transposition of the genetic elements, translocations, and alterations in microRNA expression), (2) presence of nonsensitive subpopulation (e.g., cancer stem cells) and (3) activation of mechanisms against xenobiotics (such as anticancer drugs) are responsible for reduced efficacy and treatment failure [46][47][48].
The acquired or extrinsic resistance occurs when the malignant cell line becomes less responsive to chemotherapy over time, that is, the antitumoral drug loses efficacy. The factors that lead to acquired resistance are grouped into the (4) activation of new proto-oncogenes, (5) mutations and/or alterations in transcription of drug targets, (6) modifications in the tumor microenvironment (TME) after the beginning of treatment [49][50][51].
In leukemias and lymphomas, some resistance mechanisms are more frequent than others. A study showed that the presence of cancer stem cells subpopulations in leukemia is related to less responsiveness to cytotoxicity chemotherapy, once these cells demonstrated a reduced proliferative rate [52]. In acute myeloid leukemia (AML), the treatment with the AraC is passive of acquired resistance. AraC is a pro-drug that needs to be phosphorylated to AraC-triphosphate to reach its drug target and reduced expression in metabolic pathways and mutations in AML cell lines leads to decreased levels of active drug, causing drug-resistance to AraC treatment [53][54].
Increased expression of the mechanism of drug efflux is well known as a cause of leukemia recurrence. Transmembrane transporters from the ATP Binding Cassette (ABC) superfamily ABCB1 (MDR1), ABCC1, ABCG2 are involved in the acquired resistance in AML, acute lymphocytic leukemia (ALL) and chronic myeloid leukemia (CML) [55][56][57].
CML is characterized by the presence of the Philadelphia chromosome (Ph+). The translocation between chromosomes 9 and 22 creates a chimeric protein BCR activator of RhoGEF and GTPase-ABL proto-oncogene 1 (BCR-ABL1), which has a tyrosine kinase activity [58][59]. The targeted therapy with tyrosine kinase inhibitors (TKIs) shows good therapeutical effect in CML patients, but mutations in CML patients genotypes are now the new challenge to TKIs therapy and in the CML clinic, once approximately 20–30% of patients do not respond to TKI from first-generation [58][60][61].
2. The Advent of Kinase Inhibition
Protein kinases (PK) are signaling regulators involved in various cellular functions including metabolism, cell cycle regulation, survival, and differentiation. Once activated, PKs typically phosphorylate serine, threonine or tyrosine residues on the target protein, leading to conformational change and consequent functional activation of the target proteins [62].
Phosphorylation of the target proteins by kinases is tightly regulated, and any perturbation to this regulation may lead to a diseased state. Multiple mechanisms lead to deregulation of kinases, enhancing oncogenic potential, which may include overexpression, relocation, fusions, point mutations and deregulation of upstream signaling (Figure 2). These discoveries led to the development of several KIs with wide applications in the clinical practice [62][63].
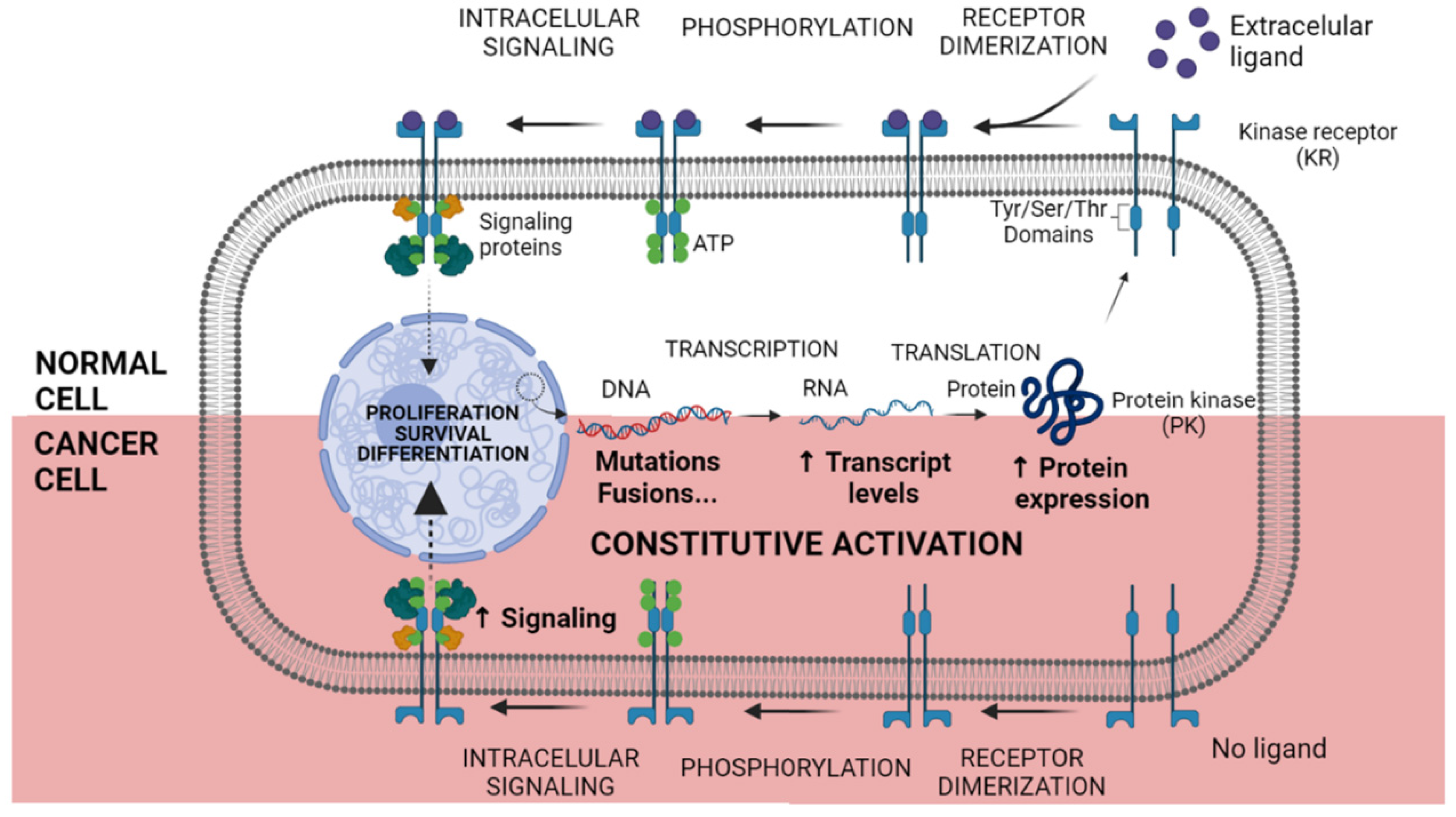
Figure 2. Constitutive activation of Kinase Receptor mechanism. In normal cells, after transcription and translation, Kinase Receptors (KR) are sent to the cellular membrane. In the presence of an extracellular ligand, the KR dimerization occurs and the Tyrosine (Tyr), Serine (Ser) or Threonine (Thr) domains are autophosphorylated. Signaling proteins are attached to the phosphorylated amino acid residues, and the downstream signalization begins. Otherwise, in cancer cells, genetic and chromosomal alterations lead to enhanced RNA and protein overexpression. Malignant cancer cells do not depend on an extracellular ligand to initiate the signaling process, being constitutively active and boosting tumoral phenotype. Created with BioRender.com, accessed date: 20 August 2021.
Imatinib was the first TKI to be approved by the US Food and Drug Administration (FDA) for the treatment of patients with CML. Imatinib inhibits the kinase activity of the fusion protein BCR-ABL1—encoded by a gene mutated in all patients with Ph+ CML—through a competitive mechanism at the ATP-binding site [64]. This blockage prevents the transduction of intracellular signals necessary for cell proliferation and apoptosis evasion. In addition, imatinib inhibits the proliferation of cells from different CML lineages and hematopoietic progenitor cells [65].
Imatinib (Glivec®) marked the beginning of the era of kinase inhibition as a reality for oncologic patients, although later on, the emergence of tumor resistance was unavoidable. After the introduction of imatinib in the early 2000s, the more selective second-generation drugs dasatinib, nilotinib, and bosutinib, followed by the third-generation compound ponatinib, enriched the therapeutic options to treat patients with Ph+ leukemias [66][67].
Approximately 20 to 30% of patients are bound to express resistance to imatinib. The mechanisms of resistance can be explained by mutations in the kinase domain of the BCR-ABL chimeric protein, gene amplification and overexpression of the BCR-ABL gene, alteration in the expression of influx and efflux transmembrane proteins and alterations in the regulation of signal transduction mechanisms. The T315I mutation in the BCR-ABL kinase domain is the most clinically alarming because only the third-generation inhibitor, ponatinib, has demonstrated efficiency in treating T315I-mutated tumors [68].
Dasatinib is a second-generation TKI approved for the treatment of CML and Ph+ ALL following imatinib treatment failure. Its range of targets include BCR-ABL, KIT proto-oncogene (c-Kit), platelet-derived growth factor receptor (PDGFR), and members of the SRC proto-oncogene (SRC) family [62][63][64]. Due to its ability to inhibit the proliferation of most mutant cells resistant to imatinib, dasatinib has been shown to be a good alternative for the treatment of patients who do not respond well to imatinib. However, a high toxicity of this drug was observed in patients in the more advanced stages of the disease due to the necessary high therapeutic doses [65].
Nilotinib is also a second-generation BCR-ABL kinase inhibitor. It has a similar spectrum of kinase targets that includes BCR-ABL, c-Kit and PDGFR and has activity against most imatinib resistance-conferring mutations, except the T315I mutation domain of the BCR-ABL gene, for which it remained ineffective. It was designed to overcome the imatinib resistance by binding to the kinase domain of imatinib-resistant mutants of BCR-ABL and imatinib-sensitive BCR-ABL with higher affinity [62][64][68].
The molecules that act as inhibitors of PKs catalytic activities are classified according to the mechanism of interaction with the kinase domain of target protein, being categorized into reversible and irreversible inhibitors. The reversible inhibitors are subclassified into types I, I½, II, III, IV or V, according to their connection and interaction with the PK domains [69].
Type I inhibitors are also known as “competitive ATP” inhibitors because they interact at the ATP binding site in the PK domain in its active conformation. The molecular recognition site of type I, I½ and II inhibitors is the hinge region. Examples of FDA-approved type I protein kinase inhibitors are gefitinib (epidermal growth factor receptor (EGFR) inhibitor), sunitinib (vascular endothelial growth factor receptor (VEGFR) and PDGFR inhibitor) and dasatinib (BCR-ABL inhibitor) [69].
Type I½ inhibitors interact with the activation segment of the kinase domain in a conformation that points towards the ATP-binding site (DFG-in) while type II inhibitors target the PK activation segment in the inactive conformation that points away from the ATP-binding site (DFG-out). Examples of FDA-approved type I½ and type II PK inhibitors are lapatinib (EGFR inhibitor) and nilotinib (inhibitor of BCR-ABL), respectively [69].
Type III and IV inhibitors are both allosteric in nature. While type III molecular recognition occurs exclusively at an allosteric site adjacent to the hinge, the type IV category refers to those allosteric inhibitors whose molecular recognition occur at a site distant from the hinge, neither exerting direct action over the ATP binding site. These inhibitors have the advantage of ensuring greater selectivity for the targeted proteins. Trametinib and cobimetinib are currently type III PK inhibitors approved by the FDA, both targeting mitogen-activated protein kinase kinase (MEK), and no type IV inhibitors have FDA approval to date. Type V inhibitors have bivalent activity, binding to two different regions of the PK domain [70].
The irreversible inhibitors are able to interact with the target protein through covalent bond formation and most recently have been categorized as type VI inhibitors. Examples of FDA-approved covalent and irreversible PK inhibitors are afatinib (erb-b2 receptor tyrosine kinase 2 (HER2) and EGFR inhibitor), ibrutinib (Bruton’s tyrosine kinase (BTK) inhibitor) and osimertinib (selective mutant T790M EGFR inhibitor) [70].
Kinase inhibition has revolutionized the practice of oncology and hematology for the past 20 years with over 40 compounds approved by the FDA. The therapeutic potential of kinase inhibition in oncology has been rapidly expanding beyond its origins in receptor tyrosine kinase oncogenes. The emergence of resistance mechanisms to existing kinase-targeting drugs has motivated a search for alternative targets, and the expansion of kinase inhibitor drugs into new target space continues to be facilitated by innovative strategies in precision medicine and molecular techniques [71][72].
3. Recent Prospects into Clinical Investigations
The usage of KIs in the treatment of several leukemia subtypes is an already established course of action in clinical practice. CML, FMS-like tyrosine kinase 3 (FLT3)-mutated AML and B-cell neoplasms are amongst the malignancies most responsive to KI treatment, including FDA approved drugs [73][74][75].
A large spectrum of different KIs and treatment protocols were covered, and kinase inhibition was mainly evaluated as a single-agent strategy in the treatment of R/R hematologic disorders, with only 10 out of 32 clinical trials associating KI usage with another cytotoxic agent [76][77][78][79][80][81][82][83][84][85][86][87][88][89][90][91][92][93][94][95][96][97][98][99][100][101][102][103][104][105][106][107] (Figure 3).
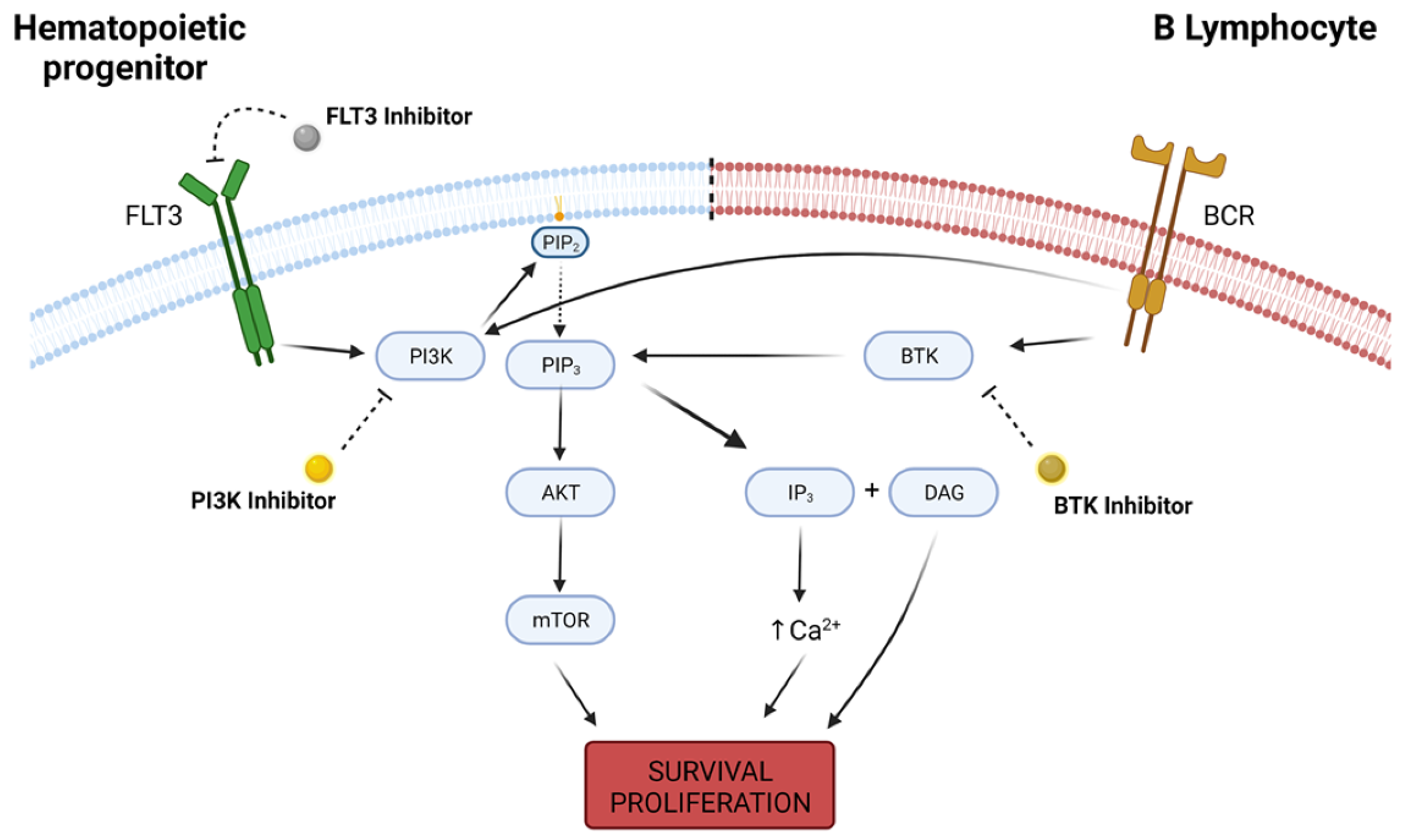
Figure 3. Intracellular pathways of FLT3, PI3K and BTK. FLT3 is a receptor tyrosine kinase highly expressed in hematopoietic progenitor cells, and its activity leads to downstream activation of survival pathways such as PI3K, which in turn converts phosphatidylinositol 4,5-bisphosphate (PIP2) into phosphatidylinositol-3,4,5-trisphosphate (PIP3) and recruits AKT to the cell membrane, with further upregulation in mTOR activity. PI3K/AKT/mTOR pathway is responsible for a wide variety of physiological functions that determine cellular survival and proliferation. On B lymphocites, B-cell receptor (BCR) is a transmembrane immunoglobulin that, upon activation, signals for PI3K and BTK activity. BTK regulates PIP3 degradation into IP3 (inositol 1-4-5 trisphosphate) and DAG (diacylglycerol), increasing intracellular calcium concentration and also promoting cellular survival and proliferation. Deregulation in the expression of any of these kinases may lead to malignant cell phenotype, and kinase inhibitors are a possible therapeutic option in the oncological practice. Created with BioRender.com, accessed date: 20 August 2021.
It is important to note that, in the analyzed studies, no clinical trial investigating KI efficiency in CML or ALL cohorts was identified. This observation is likely due to the existence of already consolidated therapy options for both malignant subtypes. While CML patients highly benefit from the usage of imatinib and other second-generation TKIs, pediatric ALL patients achieve high probabilities of survival when treated under protocols of induction and consolidation therapies utilizing cytotoxic agents, with adult ALL patients adopting pediatric-inspired treatment regimens [108][109][110].
4. AML and Myeloproliferative Disorders
FLT3 is a receptor tyrosine kinase with a major role in hematopoiesis, being expressed in undifferentiated myeloid and lymphoid progenitors. Mutations in different FLT3 domains are associated with poor prognosis in AML patients and can be expressed as either internal tandem duplication (FLT3-ITD) or tyrosine kinase domain (FLT3-TKD) mutations. Usage of FLT3 inhibitors, as a monotherapy or in combination regimens, have demonstrated superior outcomes to standard chemo-immunotherapy and are a promising prospect for the future of AML treatments, even though impairments regarding tumor-acquired resistance and duration of response to the treatment are still relevant in the clinical practice [111][112][113].
Sorafenib is an FDA approved KI for the treatment of renal cell carcinomas and hepatocellular carcinomas that possesses multikinase inhibition proprieties, with VEGFR inhibition being the main focus of interest in clinical practice [114][115]. As a type II inhibitor, sorafenib binds to the activation loop of inactive VEGFR forms in a reversible manner by interacting with hydrophobic allosteric pockets deep within the kinase structure [116].
Although not a standard in clinical practice, sorafenib has a demonstrated activity in FLT3-ITD inhibition and has been used, with modest results achieved, as a monotherapy for the treatment of AML patients in previous clinical trials [117][118]. While, in the analyzed studies, usage of sorafenib in combination therapies for the treatment of R/R myeloproliferative disorders yielded encouraging results based on response rates, parameters of patient overall survival and progression-free survival are still generally poor and may relate to inhibitor’s inability to induce deep molecular response and reduction of FLT3-ITD allelic burden in all treated patients [79][86].
Clinical trials from the past year also reassured sorafenib’s importance in AML treatment. Burchert et al. and Xuan et al. demonstrated, through individual studies, that usage of sorafenib by patients afflicted with FLT3-ITD AML after allogeneic hematopoietic stem-cell transplantation increases relapse-free survival while presenting minimal toxicities when compared to the placebo group [119][120].
Gilteritinib and quizartinib are both second-generation inhibitors, the former being a type I inhibitor and the latter being type II, with high selectivity for FLT3. While only gilteritinib has FDA approval for the treatment of R/R AML, studies utilizing either inhibitor for the treatment of AML FLT3-mutated patients have already described increased response rates over traditional salvage chemotherapy [121][122][123].
The problem around quizartinib approval for clinical use in AML management is the considerably low benefit-ratio due to the emergence of tumor TKD-mutation-mediated resistance. FLT3-TKD mutations frequently occur at the kinase activation loop and lead to constitutively active kinases which are not suitable targets for type II inhibitors because this subclass is dependent on the inactive protein conformation to be able to adequately bind around the ATP sites. As a type I inhibitor, which binds to active kinase conformations, gilteritinib is able to avoid resistance mechanisms that hinder type II inhibitor activity, and it demonstrates increased efficiency as a monotherapy in R/R AML patients [123][124][125].
Two clinical trials evaluating gilteritinib as a monotherapy reported increased treatment efficacy, which translates to higher overall response rates (ORR) and complete remissions when applied to patients whose FLT3 mutation-positive status was known compared to patients with wild type FLT3 [99][103]. Such results may be attributed to the increased specificity of gilteritinib, as a second-generation inhibitor, in targeting FLT3 when compared to KIs such as sorafenib, which, in fact, have multikinase activities and may also regulate tumorigenesis through other distinct molecular pathways [126].
Patients suffering from myeloproliferative disorders under treatment regimens that included KIs with molecular targets other than FLT3 inhibition did not report significant clinical responses and parameters of survival and disease progression were generally poor. However, the occurrence of adverse events (AE) was not drastically different from what is reported of most KI therapies, indicating a tolerable profile that may permit further studies in the investigation of their association with synergetic compounds [91][94][97][98][104][106][107].
An exception to the above-mentioned inefficacy of KIs other than FLT3 inhibitors was the activity of rigosertib, an inhibitor of RAS signaling pathways, when used alone or in combination with azacitidine, a nucleoside analog, for the treatment of myelodysplastic syndrome (MDS) patients. The ORR and bone marrow responses for this patient cohort demonstrated encouraging results, and the observed AEs did not represent an impairment for continuous therapy, even when analyzed in a drug combination regimen [80][102].
5. Lymphoid Malignancies
PI3K is a family of protein kinases that act as second messengers downstream of receptor tyrosine kinases and G-protein-coupled receptors. Their activity in cellular proliferation and metabolism is highly associated with cascade activation of AKT serine/threonine kinase (AKT)/mechanistic target of rapamycin kinase (mTOR) pathways, although PI3K AKT/mTOR-independent mechanisms are also relevant to cancer development [127][128].
While its well-defined role in carcinogenesis puts a spotlight into PI3K inhibition in the oncological practice, the many physiological cellular routes associated with PI3K/AKT/mTOR pathway represent an impairment to its proper targeting in cancer due to an elevated number of related toxicities and off-target effects [129]. A priority in the development of selective PI3K inhibitors with activity on specific kinase isoforms has been seen in the recent years over the development of pan-PI3K inhibitors, which are responsible for a broader spectrum of treatment related AEs [130].
Idelalisib is a selective PI3K-δ isoform inhibitor and was the first PI3K inhibitor to receive FDA approval, being indicated to treat R/R chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and R/R follicular lymphoma (FL) [131]. PI3K-δ is highly expressed in malignant B-cells, being associated with tumor proliferation and apoptosis evasion, and idelalisib’s inhibition over PI3K-δ regulates the downstream activities of B-cell receptor pathways, which are also main effectors in B-cell malignancies pathogenesis [132]. Other relevant PI3K inhibitors with FDA approval to treat hematological malignancies include the pan-PI3K inhibitor with preferential activity towards PI3K-α/-δ copanlisib, the PI3K-γ/-δ inhibitor duvelisib and the recently approved PI3K-δ/Casein kinase 1 epsilon (CSNK1E) inhibitor umbralisib [133][134][135].
The high prevalence of clinical trials evaluating PI3K inhibition as therapeutics for B-cell malignancies speaks to the favorable outcomes, especially when combined with chemo-immunotherapy treatment regimens, achieved in these studies, with ORRs reaching results as high as 75% of the treated population. Treatment efficacy, however, is diverse among different malignant B-cell subtypes, and patients afflicted with R/R diffuse large B-cell lymphoma (DLBCL) had generally lower rates of response to PI3K inhibition. Even among DLBCL patients, molecular profiles distinguishing the cell of origin in activated B-cell-like (ABC) DLBCL and germinal center B-cell-like (GCB) DLBCL represent a further stratification when predicting patient outcome to PI3K inhibition treatment [84][87][89][93][95][96][101][105].
Mechanisms involved in tumor-acquired PI3K-inhibitor resistance are not fully elucidated yet, with no common mutation characterized across patient cohorts with progressive disease after idelalisib treatment [136]. However, analyses in human and murine models signal towards upregulation of mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinases (ERK) pathways in neoplastic cells resistant to PI3K-δ inhibition, which are major cellular mechanisms responsible for the regulation of proliferation, differentiation and cell death [137][138][139]. In the murine model, MAPK/ERK activity was enhanced due to overexpression of insulin-like growth factor 1 receptor (IGF1R) and concomitant treatment with linsitinib, an IGF1R inhibitor, was able to overcome PI3K-δ resistance, indicating a possibility for investigation into combination treatments in the clinical practice [139].
Another highly relevant kinase in the development of B-cell neoplasms is BTK, and as such, it was also a prevalent molecular target in the aforementioned studies. BTK is a nonreceptor tyrosine kinase that is a member of the Tec protein tyrosine kinase (TEC) family and, alongside PI3K, BTK is a main effector of downstream B-cell receptor signaling pathways, playing a critical role in proliferation and metabolism of B-cells as well as in their carcinogenesis [140][141].
Ibrutinib is an oral and irreversible first-generation BTK inhibitor that revolutionized CLL treatment since its original FDA approval in 2013 [142]. Ibrutinib binds covalently to a cysteine residue (Cys481) in the BTK active domain, hindering kinase activity and also regulating downstream pathways. The bond formed to Cys481, however, is not completely selective to BTK and may promote ibrutinib’s activity over other off-target kinases, such as other members of the TEC family or EGFR, increasing the occurrence of treatment-related AEs and toxicities [143][144].
Although a standard of care in many lymphoid malignancies, primary and acquired resistances to ibrutinib treatment are still problematic in the oncologic routine. The specific C481S mutation, associated with a cysteine-to-serine exchange in BTK active domain, is well characterized as altering ibrutinib’s capability to covalently bind to BTK and handicapping its effectiveness over tumor-cell proliferation. Overexpression of distinct cell survival mechanisms, tumor microenvironment and cancer stem cell metabolism have also been indicated as possible routes for tumor resistance to ibrutinib in lymphoid neoplasms [145][146][147].
Acalabrutinib, zanubrutinib and tirabrutinib are second-generation irreversible inhibitors that present much more selective activity towards BTK than towards other TEC family kinases, with FDA approval to treat B-cell malignancies encompassing only the first two [148][149]. While clinical investigations into second-generation BTK inhibitors are prevalent, their mechanisms of action do not seem to overcome ibrutinib treatment resistance, but still present advantages when analyzing treatment-related AEs. Clinical trials comparing acalabrutinib or zanubrutinib efficacy over ibrutinib in patients afflicted with R/R lymphoid malignancies determined that, while their response rates are similar, the usage of second-generation inhibitors relate to much more tolerable toxicity profiles with special emphasis on cardiovascular tolerability [149][150][151].
First- and second-generation BTK inhibitors, as a monotherapy or in combination protocols, have been evaluated in clinical trials for the past half-decade, and the efficacy results verify them as a solid choice for the treatment of B-cell malignancies even as a second-line strategy, achieving exceedingly high response rates in most studies and allowing for long periods of progression-free survival and overall survival. These observed results are in line with what is expected from the utilization of BTK inhibitors because, although relatively recent in clinical use, their major role in lymphoid malignancies management is already a standard [78][81][83][85][88][92][95].
Both PI3K and BTK are well established molecular targets in cancer and count with FDA approved drugs for the treatment of an array of malignant subtypes, but still deal with emerging cases of drug resistance and inability to promote complete remission when used as monotherapy for R/R malignancies. In this context, a study conducted by Davids et al. investigated the outcome of patients under ibrutinib, a first-generation BTK inhibitor, and umbrasilib, a selective PI3K-δ inhibitor, combination therapy and determined that this treatment protocol is not only effective but also clinically safe and warrants further investigation to fully elucidate its potential in the clinical practice [95][152][153].
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics13101604
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2018, 68, 394–424.
- Fitzmaurice, C.; Abate, D.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdel-Rahman, O.; Abdelalim, A.; Abdoli, A.; Abdollahpour, I.; Abdulle, A.S.M.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2019, 5, 1749.
- Lewandowska, A.M.; Rudzki, M.; Rudzki, S.; Lewandowski, T.; Laskowska, B. Environmental Risk Factors for Cancer–Review Paper. Ann. Agric. Environ. Med. 2019, 26, 1–7.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674.
- Armitage, J.O.; Gascoyne, R.D.; Lunning, M.A.; Cavalli, F. Non-Hodgkin Lymphoma. Lancet 2017, 390, 298–310.
- Juliusson, G.; Hough, R. Leukemia. Prog. Tumor Res. 2016, 43, 87–100.
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular Targeted Therapy: Treating Cancer with Specificity. Eur. J. Pharmacol. 2018, 834, 188–196.
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in Kinase Drug Discovery: Targets, Indications and Inhibitor Design. Nat. Rev. Drug Discov. 2021, 2021, 1–23.
- Ramdass, B.; Chowdhary, A.; Koka, P.S. Hematological Malignancies: Disease Pathophysiology of Leukemic Stem Cells. J. Stem. Cells 2013, 8, 151–187.
- Nogueira, B.M.D.; Machado, C.B.; Montenegro, R.C.; Moraes, M.E.A.D.; Moreira-Nunes, C.A. Telomere Length and Hematological Disorders: A Review. In Vivo 2020, 34, 3093–3101.
- Harrison, C.J. Cytogenetics of Paediatric and Adolescent Acute Lymphoblastic Leukaemia. Br. J. Haematol. 2009, 144, 147–156.
- Montaño, A.; Forero-Castro, M.; Hernández-Rivas, J.-M.; García-Tuñón, I.; Benito, R. Targeted Genome Editing in Acute Lymphoblastic Leukemia: A Review. BMC Biotechnol. 2018, 18, 45.
- Pearce, L.; Morgan, L.; Lin, T.T.; Hewamana, S.; Matthews, R.J.; Deaglio, S.; Rowntree, C.; Fegan, C.; Pepper, C.; Brennan, P. Genetic Modification of Primary Chronic Lymphocytic Leukemia Cells with a Lentivirus Expressing CD38. Haematologica 2010, 95, 514–517.
- Islam, A. The Origin and Spread of Human Leukemia. Med. Hypotheses 1992, 39, 110–118.
- Beutler, E. The Treatment of Acute Leukemia: Past, Present, and Future. Leukemia 2001, 15, 658–661.
- Reinhard, E.H.; Good, J.T.; Martin, E. Chemotherapy of malignant neoplastic diseases. J. Am. Med. Assoc. 1950, 142, 383–390.
- Farber, S.; Diamond, L.K. Temporary Remissions in Acute Leukemia in Children Produced by Folic Acid Antagonist, 4-Aminopteroyl-Glutamic Acid. N. Engl. J. Med. 1948, 238, 787–793.
- Niu, C.; Yan, H.; Yu, T.; Sun, H.P.; Liu, J.X.; Li, X.S.; Wu, W.; Zhang, F.Q.; Chen, Y.; Zhou, L.; et al. Studies on Treatment of Acute Promyelocytic Leukemia with Arsenic Trioxide: Remission Induction, Follow-up, and Molecular Monitoring in 11 Newly Diagnosed and 47 Relapsed Acute Promyelocytic Leukemia Patients. Blood 1999, 94, 3315–3324.
- Hajdu, S.I.; Vadmal, M. A Note from History: Landmarks in History of Cancer, Part 6. Cancer 2013, 119, 4058–4082.
- Bernard, J.; Marie, J.; Salet, J.; Cruciani, C. Essai de Traitement Des Leucemies Aigues de l’enfance Par l’association Aminopterin-Cortison. Bull. Mem. Soc. Med. Hop. 1951, 16, 621–625.
- Gellhorn, A.; Hyman, G.A.; Ultmann, J.E. Chlorambucil in Treatment of Chronic Lymphocytic Leukemia and Certain Lymphomas. J. Am. Med. Assoc. 1956, 162, 178–183.
- Burchenal, J.H.; Murphy, M.L.; Ellison, R.R.; Sykes, M.P.; Tan, T.C.; Leone, L.A.; Karnofsky, D.A.; Craver, L.F.; Dargeon, H.W.; Rhoads, C.P. Clinical Evaluation of a New Antimetabolite, 6-Mercaptopurine, in the Treatment of Leukemia and Allied Diseases. Blood 1953, 8, 965–999.
- Coggins, P.R.; Ravdin, R.G.; Eisman, S.H. Clinical Evaluation of a New Alkylating Agent: Cytoxan (Cyclophosphamide). Cancer 1960, 13, 1254–1260.
- Noble, R.L.; Beer, C.T.; Cutts, J.H. Role of Chance Observations in Chemotherapy: Vinca Rosea. Ann. N. Y. Acad. Sci. 1958, 76, 882–894.
- Dubost, M.; Ganter, P.; Maral, R. Une Nouvel Antibiotic a Proprieties Antitumorales. C. R. Hebd. Seances Acad. Sci. 1963, 257, 1813–1818.
- Umezawa, H.; Ishizuka, M.; Maeda, K.; Takeuchi, T. Studies on Bleomycin. Cancer 1967, 20, 891–895.
- Galton, D.A. Myleran in Chronic Myeloid Leukaemia; Results of Treatment. Lancet 1953, 264, 208–213.
- Karon, M.; Freireich, E.J.; Frei, E.; Taylor, R.; Wolman, I.J.; Djerassi, I.; Lee, S.L.; Sawitsky, A.; Hananian, J.; Selawry, O.; et al. The Role of Vincristine in the Treatment of Childhood Acute Leukemia. Clin. Pharm. Ther. 1966, 7, 332–339.
- Hill, J.M.; Roberts, J.; Loeb, E.; Khan, A.; MacLellan, A.; Hill, R.W. L-Asparaginase Therapy for Leukemia and Other Malignant Neoplasms. Remission in Human Leukemia. JAMA 1967, 202, 882–888.
- Tan, C.; Tasaka, H.; Yu, K.-P.; Murphy, M.L.; Karnofsky, D.A. Daunomycin, an Antitumor Antibiotic, in the Treatment of Neoplastic Disease. Clinical Evaluation with Special Reference to Childhood Leukemia. Cancer 1967, 20, 333–353.
- Whiteside, J.A.; Philips, F.S.; Dargeon, H.W.; Burchenal, J.H. Intrathecal Amethopterin in Neurological Manifestations of Leukemia. AMA Arch. Intern. Med. 1958, 101, 279–285.
- Rundles, R.W.; Grizzle, J.; Bell, W.N.; Corley, C.C.; Frommeyer, W.B.; Greenberg, B.G.; Huguley, C.M.; Watson James, G.; Jones, R.; Larsen, W.E.; et al. Comparison of Chlorambucil and Myleran® in Chronic Lymphocytic and Granulocytic Leukemia. Am. J. Med. 1959, 27, 424–432.
- Rundles, R.W.; Laszlo, J.; Garrison, F.E.; Hobson, J.B. The Antitumor Spectrum of Cyclophosphamide. Cancer Chemother. Rep. 1962, 16, 407–411.
- George, P.; Hernandez, K.; Hustu, O.; Borella, L.; Holton, C.; Pinkel, D. A Study of “Total Therapy” of Acute Lymphocytic Leukemia in Children. J. Pediatr. 1968, 72, 399–408.
- Carbone, P.P.; Spurr, C.; Schneiderman, M.; Scotto, J.; Holland, J.F.; Shnider, B. Management of Patients with Malignant Lymphoma: A Comparative Study with Cyclophosphamide and Vinca Alkaloids. Cancer Res. 1968, 28, 811–822.
- Boiron, M.; Weil, M.; Jacquillat, C.; Tanzer, J.; Levy, D.; Sultan, C.; Bernard, J. Daunorubicin in the Treatment of Acute Myelocytic Leukaemia. Lancet 1969, 1, 330–333.
- Kersey, J.H. Fifty Years of Studies of the Biology and Therapy of Childhood Leukemia. Blood 1997, 90, 4243–4251.
- Devita, V.T.; Serpick, A.A.; Carbone, P.P. Combination Chemotherapy in the Treatment of Advanced Hodgkin’s Disease. Ann. Intern. Med. 1970, 73, 881–895.
- Moxley, J.H.; De Vita, V.T.; Brace, K.; Frei, E. Intensive Combination Chemotherapy and X-Irradiation in Hodgkin’s Disease. Cancer Res. 1967, 27, 1258–1263.
- Haghbin, M.; Tan, C.C.; Clarkson, B.D.; Miké, V.; Burchenal, J.H.; Murphy, M.L. Intensive Chemotherapy in Children with Acute Lymphoblastic Leukemia (L-2 Protocol). Cancer 1974, 33, 1491–1498.
- Aur, R.J.A.; Simone, J.V.; Hustu, H.O.; Verzosa, M.S. A Comparative Study of Central Nervous System Irradiation and Intensive Chemotherapy Early in Remission of Childhood Acute Lymphocytic Leukemia. Cancer 1972, 29, 381–391.
- Thomas, E.D.; Buckner, C.D.; Banaji, M.; Clift, R.A.; Fefer, A.; Flournoy, N.; Goodell, B.W.; Hickman, R.O.; Lerner, K.G.; Neiman, P.E.; et al. One Hundred Patients with Acute Leukemia Treated by Chemotherapy, Total Body Irradiation, and Allogeneic Marrow Transplantation. Blood 1977, 49, 511–533.
- Zhou, H.-S.; Carter, B.Z.; Andreeff, M. Bone Marrow Niche-Mediated Survival of Leukemia Stem Cells in Acute Myeloid Leukemia: Yin and Yang. Cancer Biol. Med. 2016, 13, 248–259.
- Wang, X.; Zhang, H.; Chen, X. Drug Resistance and Combating Drug Resistance in Cancer. Cancer Drug Resist. 2019, 2, 141–160.
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233.
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm Bull. 2017, 7, 339–348.
- Zhang, J.; Gu, Y.; Chen, B. Mechanisms of Drug Resistance in Acute Myeloid Leukemia. Onco. Targets Ther. 2019, 12, 1937–1945.
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L.; et al. Evolution and Impact of Subclonal Mutations in Chronic Lymphocytic Leukemia. Cell 2013, 152, 714–726.
- Tadesse, F.; Asres, G.; Abubeker, A.; Gebremedhin, A.; Radich, J. Spectrum of BCR-ABL Mutations and Treatment Outcomes in Ethiopian Imatinib-Resistant Patients With Chronic Myeloid Leukemia. JCO Glob. Oncol. 2021, 1187–1193.
- Huang, R.; Kang, Q.; Liu, H.; Li, Y. New Insights into the Molecular Resistance Mechanisms of Chronic Myeloid Leukemia. Curr. Cancer Drug Targets 2016, 16, 323–345.
- Quintás-Cardama, A.; Kantarjian, H.M.; Cortes, J.E. Mechanisms of Primary and Secondary Resistance to Imatinib in Chronic Myeloid Leukemia. Cancer Control 2009, 16, 122–131.
- Viale, A.; De Franco, F.; Orleth, A.; Cambiaghi, V.; Giuliani, V.; Bossi, D.; Ronchini, C.; Ronzoni, S.; Muradore, I.; Monestiroli, S.; et al. Cell-Cycle Restriction Limits DNA Damage and Maintains Self-Renewal of Leukaemia Stem Cells. Nature 2009, 457, 51–56.
- Michael, M.; Doherty, M.M. Tumoral Drug Metabolism: Overview and Its Implications for Cancer Therapy. J. Clin. Oncol. 2005, 23, 205–229.
- Wolach, O.; Itchaki, G.; Bar-Natan, M.; Yeshurun, M.; Ram, R.; Herscovici, C.; Shpilberg, O.; Douer, D.; Tallman, M.S.; Raanani, P. High-Dose Cytarabine as Salvage Therapy for Relapsed or Refractory Acute Myeloid Leukemia--Is More Better or More of the Same? Hematol. Oncol. 2016, 34, 28–35.
- Pinto, C.A.; DE Sousa Portilho, A.J.; Barbosa, M.C.; de Moraes, M.E.A.; de Lemos, J.A.R.; Burbano, R.M.R.; Moreira-Nunes, C.A. Combined Therapy of ATRA and Imatinib Mesylate Decreases BCR-ABL and ABCB1/MDR1 Expression Through Cellular Differentiation in a Chronic Myeloid Leukemia Model. In Vivo 2021, 35, 2661–2667.
- Baudis, M.; Prima, V.; Tung, Y.H.; Hunger, S.P. ABCB1 Over-Expression and Drug-Efflux in Acute Lymphoblastic Leukemia Cell Lines with t(17;19) and E2A-HLF Expression. Pediatr. Blood Cancer 2006, 47, 757–764.
- Shaffer, B.C.; Gillet, J.-P.; Patel, C.; Baer, M.R.; Bates, S.E.; Gottesman, M.M. Drug Resistance: Still a Daunting Challenge to the Successful Treatment of AML. Drug Resist. Updates 2012, 15, 62–69.
- Eadie, L.N.; Hughes, T.P.; White, D.L. ABCB1 Overexpression Is a Key Initiator of Resistance to Tyrosine Kinase Inhibitors in CML Cell Lines. PLoS ONE 2016, 11, e0161470.
- Machado, C.B.; Nogueira, B.M.D.; de Sousa Portilho, A.J.; de Moraes Filho, M.O.; de Moraes, M.E.A.; de Fátima Aquino Moreira Nunes, C. Caracterização do Cromossomo Philadephia em Tumores Não-Sólidos: Uma Abordagem Citogenética Ao Câncer. In A Genética e a Construção de Novos Paradigmas Nas Ciências da Vida; Atenas: Ponta Grossa, Brazil, 2021; pp. 14–21.
- Manouchehri, A.; Kanu, E.; Mauro, M.J.; Aday, A.W.; Lindner, J.R.; Moslehi, J. Tyrosine Kinase Inhibitors (TKI) in Leukemia and Cardiovascular Events-From Mechanism to Patient Care. Arter. Thromb. Vasc. Biol. 2020, 40, 301–308.
- Yaghmaie, M.; Yeung, C.C. Molecular Mechanisms of Resistance to Tyrosine Kinase Inhibitors. Curr. Hematol. Malig. Rep. 2019, 14, 395–404.
- Kannaiyan, R.; Mahadevan, D. A Comprehensive Review of Protein Kinase Inhibitors for Cancer Therapy. Expert Rev. Anticancer Ther. 2018, 18, 1249–1270.
- Chaar, M.; Kamta, J.; Ait-Oudhia, S. Mechanisms, Monitoring, and Management of Tyrosine Kinase Inhibitors–Associated Cardiovascular Toxicities. OncoTargets Ther. 2018, 11, 6227.
- Rosti, G.; Castagnetti, F.; Gugliotta, G.; Baccarani, M. Tyrosine Kinase Inhibitors in Chronic Myeloid Leukaemia: Which, When, for Whom? Nat. Rev. Clin. Oncol. 2017, 14, 141–154.
- Pophali, P.A.; Patnaik, M.M. The Role of New Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia. Cancer J. (Sudbury Mass.) 2016, 22, 40.
- Waller, C. Imatinib Mesylate. Recent Results Cancer Res. 2018, 212, 1–27.
- Rea, D.; Ame, S.; Berger, M.; Cayuela, J.-M.; Charbonnier, A.; Coiteux, V.; Cony-Makhoul, P.; Dubruille, V.; Dulucq, S.; Etienne, G.; et al. Discontinuation of Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia: Recommendations for Clinical Practice from the French Chronic Myeloid Leukemia Study Group. Cancer 2018, 124, 2956–2963.
- de Azevedo, L.D.; Bastos, M.M.; de Oliveira, A.P.; Boechat, N. Syntheses and properties of drugs inhibitors of bcr-abl tyrosine kinase, used in the treatment of chronic myeloid leukemia. Química Nova 2017, 40, 791–809.
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2021 Update. Pharmacol. Res. 2021, 165, 105463.
- Martinez, R., III.; Defnet, A.; Shapiro, P. Avoiding or Co-Opting ATP Inhibition: Overview of Type III, IV, V, and VI Kinase Inhibitors. Next Gener. Kinase Inhib. 2020, 29.
- Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731.
- Ferguson, F.M.; Gray, N.S. Kinase Inhibitors: The Road Ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377.
- Antar, A.; Otrock, Z.; Jabbour, E.; Mohty, M.; Bazarbachi, A. FLT3 Inhibitors in Acute Myeloid Leukemia: Ten Frequently Asked Questions. Leukemia 2020, 34, 682–696.
- Singh, S.P.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s Tyrosine Kinase in B Cells and Malignancies. Mol. Cancer 2018, 17.
- Lee, H.; Basso, I.; Kim, D. Target Spectrum of the BCR-ABL Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia. Int. J. Hematol. 2021, 113, 632–641.
- Tiacci, E.; De Carolis, L.; Simonetti, E.; Capponi, M.; Ambrosetti, A.; Lucia, E.; Antolino, A.; Pulsoni, A.; Ferrari, S.; Zinzani, P.L.; et al. Vemurafenib plus Rituximab in Refractory or Relapsed Hairy-Cell Leukemia. N. Engl. J. Med. 2021, 384, 1810–1823.
- Iida, S.; Sunami, K.; Minami, H.; Hatake, K.; Sekiguchi, R.; Natsume, K.; Ishikawa, N.; Rinne, M.; Taniwaki, M. A Phase I, Dose-Escalation Study of Oral PIM447 in Japanese Patients with Relapsed and/or Refractory Multiple Myeloma. Int. J. Hematol. 2021, 113, 797–806.
- Tam, C.S.; Quach, H.; Nicol, A.; Badoux, X.; Rose, H.; Miles Prince, H.; Leahy, M.F.; Eek, R.; Wickham, N.; Patil, S.S.; et al. Zanubrutinib (BGB-3111) plus Obinutuzumab in Patients with Chronic Lymphocytic Leukemia and Follicular Lymphoma. Blood Adv. 2020, 4, 4802–4811.
- Borthakur, G.; Zeng, Z.; Cortes, J.E.; Chen, H.C.; Huang, X.; Konopleva, M.; Ravandi, F.; Kadia, T.; Patel, K.P.; Daver, N.; et al. Phase 1 Study of Combinatorial Sorafenib, G-CSF, and Plerixafor Treatment in Relapsed/Refractory, FLT3-ITD-Mutated Acute Myelogenous Leukemia Patients. Am. J. Hematol. 2020, 95, 1296–1303.
- Navada, S.C.; Garcia-Manero, G.; OdchimarReissig, R.; Pemmaraju, N.; Alvarado, Y.; Ohanian, M.N.; John, R.B.; Demakos, E.P.; Zbyszewski, P.S.; Maniar, M.; et al. Rigosertib in Combination with Azacitidine in Patients with Myelodysplastic Syndromes or Acute Myeloid Leukemia: Results of a Phase 1 Study. Leuk. Res. 2020, 94.
- Xu, W.; Yang, S.; Zhou, K.; Pan, L.; Li, Z.; Zhou, J.; Gao, S.; Zhou, D.; Hu, J.; Feng, R.; et al. Treatment of Relapsed/Refractory Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma with the BTK Inhibitor Zanubrutinib: Phase 2, Single-Arm, Multicenter Study. J. Hematol. Oncol. 2020, 13.
- Smith, C.C.; Levis, M.J.; Frankfurt, O.; Pagel, J.M.; Roboz, G.J.; Stone, R.M.; Wang, E.S.; Severson, P.L.; West, B.L.; Le, M.H.; et al. A Phase 1/2 Study of the Oral FLT3 Inhibitor Pexidartinib in Relapsed/ Refractory FLT3-ITD–Mutant Acute Myeloid Leukemia. Blood Adv. 2020, 4, 1711–1721.
- Sun, C.; Nierman, P.; Kendall, E.K.; Cheung, J.; Gulrajani, M.; Herman, S.E.M.; Pleyer, C.; Ahn, I.E.; Stetler-Stevenson, M.; Yuan, C.M.; et al. Clinical and Biological Implications of Target Occupancy in CLL Treated with the BTK Inhibitor Acalabrutinib. Blood 2020, 136, 93–105.
- Lenz, G.; Hawkes, E.; Verhoef, G.; Haioun, C.; Thye Lim, S.; Seog Heo, D.; Ardeshna, K.; Chong, G.; Haaber, J.; Shi, W.; et al. Single-Agent Activity of Phosphatidylinositol 3-Kinase Inhibition with Copanlisib in Patients with Molecularly Defined Relapsed or Refractory Diffuse Large B-Cell Lymphoma. Leukemia 2020, 34, 2184–2197.
- Byrd, J.C.; Wierda, W.G.; Schuh, A.; Devereux, S.; Chaves, J.M.; Brown, J.R.; Hillmen, P.; Martin, P.; Awan, F.T.; Stephens, D.M.; et al. Acalabrutinib Monotherapy in Patients with Relapsed/ Refractory Chronic Lymphocytic Leukemia: Updated Phase 2 Results. Blood 2020, 135, 1204–1213.
- Zhang, C.; Lam, S.S.Y.; Leung, G.M.K.; Tsui, S.P.; Yang, N.; Ng, N.K.L.; Ip, H.W.; Au, C.H.; Chan, T.L.; Ma, E.S.K.; et al. Sorafenib and Omacetaxine Mepesuccinate as a Safe and Effective Treatment for Acute Myeloid Leukemia Carrying Internal Tandem Duplication of Fms-like Tyrosine Kinase 3. Cancer 2020, 126, 344–353.
- Lunning, M.; Vose, J.; Nastoupil, L.; Fowler, N.; Burger, J.A.; Wierda, W.G.; Schreeder, M.T.; Siddiqi, T.; Flowers, C.R.; Cohen, J.B.; et al. Ublituximab and Umbralisib in Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma and Chronic Lymphocytic Leukemia. Blood 2019, 134, 1811–1820.
- Munir, T.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Barr, P.M.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Final Analysis from RESONATE: Up to Six Years of Follow-up on Ibrutinib in Patients with Previously Treated Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma. Am. J. Hematol. 2019, 94, 1353–1363.
- Flinn, I.W.; Cherry, M.A.; Maris, M.B.; Matous, J.V.; Berdeja, J.G.; Patel, M. Combination Trial of Duvelisib (IPI-145) with Rituximab or Bendamustine/Rituximab in Patients with Non-Hodgkin Lymphoma or Chronic Lymphocytic Leukemia. Am. J. Hematol. 2019, 94, 1325–1334.
- Takahashi, T.; Usuki, K.; Matsue, K.; Ohno, H.; Sakura, T.; Imanaka, R.; Murakami, M.; Ohwada, S.; Takagi, T.; Sakajiri, S. Efficacy and Safety of Quizartinib in Japanese Patients with FLT3-ITD Positive Relapsed or Refractory Acute Myeloid Leukemia in an Open-Label, Phase 2 Study. Int. J. Hematol. 2019, 110, 665–674.
- Kessler, T.; Koschmieder, S.; Schliemann, C.; Crysandt, M.; Mikesch, J.H.; von Stillfried, S.; Stelljes, M.; Pohlen, M.; Lenz, G.; Kirsch, A.; et al. Phase II Clinical Trial of Pazopanib in Patients with Acute Myeloid Leukemia (AML), Relapsed or Refractory or at Initial Diagnosis without an Intensive Treatment Option (PazoAML). Ann. Hematol. 2019, 98, 1393–1401.
- Munakata, W.; Ando, K.; Hatake, K.; Fukuhara, N.; Kinoshita, T.; Fukuhara, S.; Shirasugi, Y.; Yokoyama, M.; Ichikawa, S.; Ohmachi, K.; et al. Phase I Study of Tirabrutinib (ONO-4059/GS-4059) in Patients with Relapsed or Refractory B-Cell Malignancies in Japan. Cancer Sci. 2019, 110, 1686–1694.
- Forero-Torres, A.; Ramchandren, R.; Yacoub, A.; Wertheim, M.S.; Edenfield, W.J.; Caimi, P.; Gutierrez, M.; Akard, L.; Escobar, C.; Call, J.; et al. Parsaclisib, a Potent and Highly Selective PI3Kd Inhibitor, in Patients with Relapsed or Refractory B-Cell Malignancies. Blood 2019, 133, 1742–1752.
- Maiti, A.; Naqvi, K.; Kadia, T.M.; Borthakur, G.; Takahashi, K.; Bose, P.; Daver, N.G.; Patel, A.; Alvarado, Y.; Ohanian, M.; et al. Phase II Trial of MEK Inhibitor Binimetinib (MEK162) in RAS-Mutant Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2019, 19, 142–148.
- Davids, M.S.; Kim, H.T.; Nicotra, A.; Savell, A.; Francoeur, K.; Hellman, J.M.; Bazemore, J.; Miskin, H.P.; Sportelli, P.; Stampleman, L.; et al. Umbralisib in Combination with Ibrutinib in Patients with Relapsed or Refractory Chronic Lymphocytic Leukaemia or Mantle Cell Lymphoma: A Multicentre Phase 1-1b Study. Lancet Haematol. 2019, 6, e38–e47.
- Flinn, I.W.; Hillmen, P.; Montillo, M.; Nagy, Z.; Illés, Á.; Etienne, G.; Delgado, J.; Kuss, B.J.; Tam, C.S.; Gasztonyi, Z.; et al. The Phase 3 DUO Trial: Duvelisib vs Ofatumumab in Relapsed and Refractory CLL/SLL. Blood 2018, 132, 2446–2455.
- Abaza, Y.; Hidalgo-Lopez, J.E.; Verstovsek, S.; Jabbour, E.; Ravandi, F.; Borthakur, G.; Estrov, Z.; Alvarado, Y.; Burger, J.; Schneider, H.; et al. Phase I Study of Ruxolitinib in Previously Treated Patients with Low or Intermediate-1 Risk Myelodysplastic Syndrome with Evidence of NF-KB Activation. Leuk. Res. 2018, 73, 78–85.
- Dalle, I.A.; Cortes, J.E.; Pinnamaneni, P.; Lamothe, B.; Duque, A.D.; Randhawa, J.; Pemmaraju, N.; Jabbour, E.; Ferrajoli, A.; Wierda, W.G.; et al. A Pilot Phase II Study of Erlotinib for the Treatment of Patients with Relapsed/Refractory Acute Myeloid Leukemia. Acta Haematol. 2018, 140, 30–39.
- Usuki, K.; Sakura, T.; Kobayashi, Y.; Miyamoto, T.; Iida, H.; Morita, S.; Bahceci, E.; Kaneko, M.; Kusano, M.; Yamada, S.; et al. Clinical Profile of Gilteritinib in Japanese Patients with Relapsed/Refractory Acute Myeloid Leukemia: An Open-Label Phase 1 Study. Cancer Sci. 2018, 109, 3235–3244.
- Cortes, J.E.; Tallman, M.S.; Schiller, G.J.; Trone, D.; Gammon, G.; Goldberg, S.L.; Perl, A.E.; Marie, J.P.; Martinelli, G.; Kantarjian, H.M.; et al. Phase 2b Study of 2 Dosing Regimens of Quizartinib Monotherapy in FLT3-ITD–Mutated, Relapsed or Refractory AML. Blood 2018, 132, 598–607.
- Burris, H.A.; Flinn, I.W.; Patel, M.R.; Fenske, T.S.; Deng, C.; Brander, D.M.; Gutierrez, M.; Essell, J.H.; Kuhn, J.G.; Miskin, H.P.; et al. Umbralisib, a Novel PI3Kδ and Casein Kinase-1ε Inhibitor, in Relapsed or Refractory Chronic Lymphocytic Leukaemia and Lymphoma: An Open-Label, Phase 1, Dose-Escalation, First-in-Human Study. Lancet Oncol. 2018, 19, 486–496.
- Navada, S.C.; Fruchtman, S.M.; Odchimar-Reissig, R.; Demakos, E.P.; Petrone, M.E.; Zbyszewski, P.S.; Holland, J.F.; Silverman, L.R. A Phase 1/2 Study of Rigosertib in Patients with Myelodysplastic Syndromes (MDS) and MDS Progressed to Acute Myeloid Leukemia. Leuk. Res. 2018, 64, 10–16.
- Perl, A.E.; Altman, J.K.; Cortes, J.; Smith, C.; Litzow, M.; Baer, M.R.; Claxton, D.; Erba, H.P.; Gill, S.; Goldberg, S.; et al. Selective Inhibition of FLT3 by Gilteritinib in Relapsed or Refractory Acute Myeloid Leukaemia: A Multicentre, First-in-Human, Open-Label, Phase 1–2 Study. Lancet Oncol. 2017, 18, 1061–1075.
- Kantarjian, H.M.; Schuster, M.W.; Jain, N.; Advani, A.; Jabbour, E.; Gamelin, E.; Rasmussen, E.; Juan, G.; Anderson, A.; Chow, V.F.; et al. A Phase 1 Study of AMG 900, an Orally Administered Pan-Aurora Kinase Inhibitor, in Adult Patients with Acute Myeloid Leukemia. Am. J. Hematol. 2017, 92, 660–667.
- Jones, J.A.; Robak, T.; Brown, J.R.; Awan, F.T.; Badoux, X.; Coutre, S.; Loscertales, J.; Taylor, K.; Vandenberghe, E.; Wach, M.; et al. Efficacy and Safety of Idelalisib in Combination with Ofatumumab for Previously Treated Chronic Lymphocytic Leukaemia: An Open-Label, Randomised Phase 3 Trial. Lancet Haematol. 2017, 4, e114–e126.
- Schliemann, C.; Gerss, J.; Wiebe, S.; Mikesch, J.H.; Knoblauch, N.; Sauer, T.; Angenendt, L.; Kewitz, T.; Urban, M.; Butterfass-Bahloul, T.; et al. A Phase I Dose Escalation Study of the Triple Angiokinase Inhibitor Nintedanib Combined with Low-Dose Cytarabine in Elderly Patients with Acute Myeloid Leukemia. PLoS ONE 2016, 11, e0164499.
- Yee, K.W.L.; Chen, H.W.T.; Hedley, D.W.; Chow, S.; Brandwein, J.; Schuh, A.C.; Schimmer, A.D.; Gupta, V.; Sanfelice, D.; Johnson, T.; et al. A Phase I Trial of the Aurora Kinase Inhibitor, ENMD-2076, in Patients with Relapsed or Refractory Acute Myeloid Leukemia or Chronic Myelomonocytic Leukemia. Investig. New Drugs 2016, 34, 614–624.
- Jabbour, E.; Kantarjian, H. Chronic Myeloid Leukemia: 2018 Update on Diagnosis, Therapy and Monitoring. Am. J. Hematol. 2018, 93, 442–459.
- Kato, M.; Manabe, A. Treatment and Biology of Pediatric Acute Lymphoblastic Leukemia. Pediatr. Int. Off. J. Jpn. Pediatr. Soc. 2018, 60, 4–12.
- Aldoss, I.; Stein, A. Advances in Adult Acute Lymphoblastic Leukemia Therapy. Leuk. Lymphoma 2018, 59, 1033–1050.
- Lagunas-Rangel, F.; Chávez-Valencia, V. FLT3-ITD and Its Current Role in Acute Myeloid Leukaemia. Med. Oncol. (Northwood Lond. Engl.) 2017, 34.
- Kiyoi, H.; Kawashima, N.; Ishikawa, Y. FLT3 Mutations in Acute Myeloid Leukemia: Therapeutic Paradigm beyond Inhibitor Development. Cancer Sci. 2020, 111, 312.
- Eguchi, M.; Minami, Y.; Kuzume, A.; Chi, S. Mechanisms Underlying Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia. Biomedicines 2020, 8, 245.
- Garten, A.; Grohmann, T.; Kluckova, K.; Lavery, G.G.; Kiess, W.; Penke, M. Sorafenib-Induced Apoptosis in Hepatocellular Carcinoma Is Reversed by SIRT1. Int. J. Mol. Sci. 2019, 20, 4048.
- Abdelgalil, A.A.; Alkahtani, H.M.; Al-Jenoobi, F.I. Sorafenib. Profiles Drug Subst. Excip. Relat. Methodol. 2019, 44, 239–266.
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-Approved Small-Molecule Kinase Inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439.
- Zhang, W.; Konopleva, M.; Shi, Y.; McQueen, T.; Harris, D.; Ling, X.; Estrov, Z.; Quintás-Cardama, A.; Small, D.; Cortes, J.; et al. Mutant FLT3: A Direct Target of Sorafenib in Acute Myelogenous Leukemia. JNCI J. Natl. Cancer Inst. 2008, 100, 184–198.
- Borthakur, G.; Kantarjian, H.; Ravandi, F.; Zhang, W.; Konopleva, M.; Wright, J.J.; Faderl, S.; Verstovsek, S.; Mathews, S.; Andreeff, M.; et al. Phase I Study of Sorafenib in Patients with Refractory or Relapsed Acute Leukemias. Haematologica 2011, 96, 62.
- Burchert, A.; Bug, G.; Fritz, L.V.; Finke, J.; Stelljes, M.; Röllig, C.; Wollmer, E.; Wäsch, R.; Bornhäuser, M.; Berg, T.; et al. Sorafenib Maintenance After Allogeneic Hematopoietic Stem Cell Transplantation for Acute Myeloid Leukemia With FLT3–Internal Tandem Duplication Mutation (SORMAIN). J. Clin. Oncol. 2020, 38, 2993–3002.
- Xuan, L.; Wang, Y.; Huang, F.; Fan, Z.; Xu, Y.; Sun, J.; Xu, N.; Deng, L.; Li, X.; Liang, X.; et al. Sorafenib Maintenance in Patients with FLT3-ITD Acute Myeloid Leukaemia Undergoing Allogeneic Haematopoietic Stem-Cell Transplantation: An Open-Label, Multicentre, Randomised Phase 3 Trial. Lancet Oncol. 2020, 21, 1201–1212.
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740.
- Cortes, J.E.; Khaled, S.; Martinelli, G.; Perl, A.E.; Ganguly, S.; Russell, N.; Krämer, A.; Dombret, H.; Hogge, D.; Jonas, B.A.; et al. Quizartinib versus Salvage Chemotherapy in Relapsed or Refractory FLT3-ITD Acute Myeloid Leukaemia (QuANTUM-R): A Multicentre, Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet Oncol. 2019, 20, 984–997.
- Garcia-Horton, A.; Yee, K.W. Quizartinib for the Treatment of Acute Myeloid Leukemia. Expert Opin. Pharmacother. 2020, 21, 2077–2090.
- Tarver, T.C.; Hill, J.E.; Rahmat, L.; Perl, A.E.; Bahceci, E.; Mori, K.; Smith, C.C. Gilteritinib Is a Clinically Active FLT3 Inhibitor with Broad Activity against FLT3 Kinase Domain Mutations. Blood Adv. 2020, 4, 514.
- Roskoski, R. Classification of Small Molecule Protein Kinase Inhibitors Based upon the Structures of Their Drug-Enzyme Complexes. Pharmacol. Res. 2016, 103, 26–48.
- Wu, M.; Li, C.; Zhu, X. FLT3 Inhibitors in Acute Myeloid Leukemia. J. Hematol. Oncol. 2018, 11.
- Lien, E.C.; Dibble, C.C.; Toker, A. PI3K Signaling in Cancer: Beyond AKT. Curr. Opin. Cell Biol. 2017, 45, 62.
- Aoki, M.; Fujishita, T. Oncogenic Roles of the PI3K/AKT/MTOR Axis. Curr. Top. Microbiol. Immunol. 2017, 407, 153–189.
- Arafeh, R.; Samuels, Y. PIK3CA in Cancer: The Past 30 Years. Semin. Cancer Biol. 2019, 59, 36–49.
- Ellis, H.; Ma, C.X. PI3K Inhibitors in Breast Cancer Therapy. Curr. Oncol. Rep. 2019, 21, 12.
- Gilead Sciences. ZYDELIG® (Idelalisib) Tablets, for Oral Use. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/205858lbl.pdf (accessed on 21 August 2021).
- Zirlik, K.; Veelken, H. Idelalisib. Recent Results Cancer Res. 2018, 212, 243–264.
- Bayer. ALIQOPATM (Copanlisib) for Injection, for Intravenous Use. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209936s000lbl.pdf (accessed on 21 August 2021).
- Verastem, Inc. COPIKTRA (Duvelisib), Capsules for Oral Use. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/211155s000lbl.pdf (accessed on 21 August 2021).
- TG Therapeutics. UKONIQTM (Umbralisib) Tablets, for Oral Use. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213176s000lbl.pdf (accessed on 21 August 2021).
- Ghia, P.; Ljungström, V.; Tausch, E.; Agathangelidis, A.; Scheffold, A.; Scarfo, L.; Jebaraj, B.M.C.; Owen, C.J.; Barrientos, J.C.; Zapatka, M.; et al. Whole-Exome Sequencing Revealed No Recurrent Mutations within the PI3K Pathway in Relapsed Chronic Lymphocytic Leukemia Patients Progressing Under Idelalisib Treatment. Blood 2016, 128, 2770.
- Ondrisova, L.; Mraz, M. Genetic and Non-Genetic Mechanisms of Resistance to BCR Signaling Inhibitors in B Cell Malignancies. Front. Oncol. 2020, 10.
- Murali, I.; Kasar, S.; McWilliams, E.M.; Itchaki, G.; Tyekucheva, S.; Livitz, D.; Leshchiner, I.; Dong, S.; Fernandes, S.M.; Getz, G.; et al. Activating MAPK Pathway Mutations Mediate Primary Resistance to PI3K Inhibitors in Chronic Lymphocytic Leukemia (CLL). Blood 2018, 132, 587.
- Scheffold, A.; Jebaraj, B.M.C.; Tausch, E.; Bloehdorn, J.; Ghia, P.; Yahiaoui, A.; Dolnik, A.; Blätte, T.J.; Bullinger, L.; Dheenadayalan, R.P.; et al. IGF1R as Druggable Target Mediating PI3K-δ Inhibitor Resistance in a Murine Model of Chronic Lymphocytic Leukemia. Blood 2019, 134, 534–547.
- Wang, X.; Kokabee, L.; Kokabee, M.; Conklin, D.S. Bruton’s Tyrosine Kinase and Its Isoforms in Cancer. Front. Cell Dev. Biol. 2021, 9.
- Molina-Cerrillo, J.; Alonso-Gordoa, T.; Gajate, P.; Grande, E. Bruton’s Tyrosine Kinase (BTK) as a Promising Target in Solid Tumors. Cancer Treat. Rev. 2017, 58, 41–50.
- Pharmacyclics. IMBRUVICA® (Ibrutinib) Capsules, for Oral Use. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210563s000lbl.pdf (accessed on 21 August 2021).
- Deeks, E. Ibrutinib: A Review in Chronic Lymphocytic Leukaemia. Drugs 2017, 77, 225–236.
- Zi, F.; Yu, L.; Shi, Q.; Tang, A.; Cheng, J. Ibrutinib in CLL/SLL: From Bench to Bedside (Review). Oncol. Rep. 2019, 42, 2213–2227.
- Woyach, J.A.; Furman, R.R.; Liu, T.-M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.-H.; Steggerda, S.M.; Versele, M.; et al. Resistance Mechanisms for the Bruton’s Tyrosine Kinase Inhibitor Ibrutinib. N. Engl. J. Med. 2014, 370, 2286.
- Furman, R.R.; Cheng, S.; Lu, P.; Setty, M.; Perez, A.R.; Guo, A.; Racchumi, J.; Xu, G.; Wu, H.; Ma, J.; et al. Ibrutinib Resistance in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2014, 370, 2352.
- George, B.; Chowdhury, S.M.; Hart, A.; Sircar, A.; Singh, S.K.; Nath, U.K.; Mamgain, M.; Singhal, N.K.; Sehgal, L.; Jain, N. Ibrutinib Resistance Mechanisms and Treatment Strategies for B-Cell Lymphomas. Cancers 2020, 12, 1328.
- Wu, J.; Liu, C.; Tsui, S.T.; Liu, D. Second-Generation Inhibitors of Bruton Tyrosine Kinase. J. Hematol. Oncol. 2016, 9.
- Bond, D.A.; Woyach, J.A. Targeting BTK in CLL: Beyond Ibrutinib. Curr. Hematol. Malig. Rep. 2019, 14, 197–205.
- Byrd, J.C.; Hillmen, P.; Ghia, P.; Kater, A.P.; Chanan-Khan, A.A.A.; Furman, R.R.; O’Brien, S.M.; Yenerel, M.N.; Illés, Á.; Kay, N.E.; et al. First Results of a Head-to-Head Trial of Acalabrutinib versus Ibrutinib in Previously Treated Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2021, 39, 7500.
- Tam, C.S.; Opat, S.; D’Sa, S.; Jurczak, W.; Lee, H.-P.; Cull, G.; Owen, R.G.; Marlton, P.; Wahlin, B.E.; Sanz, R.G.; et al. A Randomized Phase 3 Trial of Zanubrutinib vs Ibrutinib in Symptomatic Waldenström Macroglobulinemia: The ASPEN Study. Blood 2020, 136, 2038.
- Brullo, C.; Villa, C.; Tasso, B.; Russo, E.; Spallarossa, A. Btk Inhibitors: A Medicinal Chemistry and Drug Delivery Perspective. Int. J. Mol. Sci. 2021, 22, 7641.
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in Cancer: Mechanisms and Advances in Clinical Trials. Mol. Cancer 2019, 18.
This entry is offline, you can click here to edit this entry!