Spinal muscular atrophy (SMA), the main genetic cause of infant death, is a neurodegenerative disease characterized by the selective loss of motor neurons in the anterior horn of the spinal cord, accompanied by muscle wasting. Pathomechanically, SMA is caused by low levels of the survival motor neuron protein (SMN) resulting from the loss of the SMN1 gene. However, emerging research extends the pathogenic effect of SMN deficiency beyond motor neurons. A variety of metabolic abnormalities, especially altered fatty acid metabolism and impaired glucose tolerance, has been described in isolated cases of SMA; therefore, the impact of SMN deficiency in metabolic abnormalities has been speculated. Although the life expectancy of these patients has increased due to novel disease-modifying therapies and standardization of care, understanding of the involvement of metabolism and nutrition in SMA is still limited. Optimal nutrition support and metabolic monitoring are essential for patients with SMA, and a comprehensive nutritional assessment can guide personalized nutritional therapy for this vulnerable population. It has recently been suggested that metabolomics studies before and after the onset of SMA in patients can provide valuable information about the direct or indirect effects of SMN deficiency on metabolic abnormalities. Furthermore, identifying and quantifying the specific metabolites in SMA patients may serve as an authentic biomarker or therapeutic target for SMA.
- spinal muscular atrophy
- metabolomics
- nutrition
- therapeutics
- biomarkers
1. Introduction
Spinal muscular atrophy (SMA) is a congenital neuromuscular disease characterized by progressive muscle weakness resulting from the degeneration of motor neurons (MN) in the spinal cord [1]. Although SMA is considered a rare disease and the global incidence of live births is estimated to be about 1/10,000, SMA is still the second most common autosomal recessive genetic disease and the most common monogenic disorder that causes early infant death [2]. The carrier frequency varies from 1 in 38 to 1 in 72 among different ethnic groups, with a pan-ethnic average of 1 in 54 [3][4].
In a pathological view, SMA is resulted from an insufficient level of a 38 kDa protein, called the survival motor neuron (SMN), as a result of homologous deletion or mutation of the Survival of Motor Neuron 1 (SMN1) gene [5]. Subsequent studies showed that two genes encode SMN protein in humans: SMN1 and a 99% identical copy in sequence, known as SMN2. Indeed, SMN2 mainly differs from SMN1 by a single nucleotide (C-to-T) substitution in the exon 7 [6]. Such a critical variant results in exon 7 exclusion in most transcripts (90%) of SMN2, SMN∆7. Unlike the SMN1 gene, SMN2 can only produce 10 % full-length (FL) SMN [7]. Given that the residual FL-SMN2 transcripts can compensate for defect SMN1 to a limited extent, the SMA severity is partially rescued by SMN2 copy numbers [8]. However, the correlation between this phenotype and genotype is not absolute, and recent studies have pointed out that other potential cellular mechanisms may also be involved in modifying the clinical severity of SMA [9].
It is still unclear whether the pathogenesis of SMA is caused by a specific pattern or a combination of dysregulated effects. The cell-autonomous effects due to SMN deficiency are the main causes of MN degeneration; however, it cannot be explained for the full SMA phenotype, implicating not only dysregulated neural networks but other non-neuronal cell types involved in the SMA pathology [10][11]. Emerging research extends the pathogenic effect of SMN deficiency beyond the MN, including other cells inside and outside the central nervous system, so that many peripheral organs and non-neural tissues show pathological changes in preclinical SMA models and diseased patients (Figure 1) [12][13][14]. Furthermore, increasing evidence suggests metabolic abnormalities in patients with SMA, such as altered fatty acid metabolism, impaired glucose tolerance, and muscle mitochondria defects [15][16][17]. Recent studies also indicate that many SMA patients are either undernourished, underfed, or overfed [18]. Notably, in some SMA patients, metabolic dysregulations may even present before their first neuromuscular signs [19]. These findings suggest that SMN is essential for the survival of motor neurons and affects certain enzyme production in the metabolism.
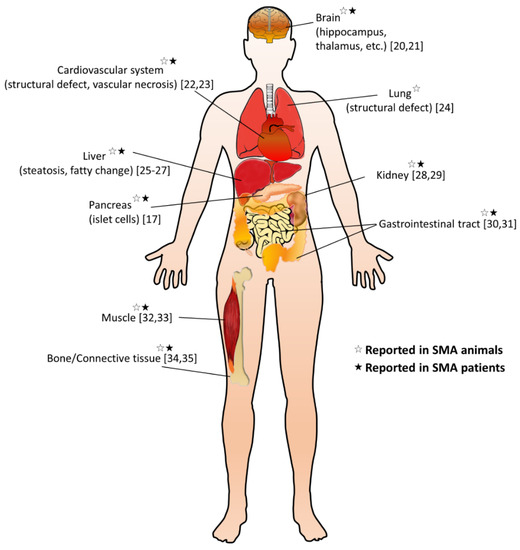
Figure 1. Overview of non-neuromuscular systemic pathology in spinal muscular atrophy (SMA). A summary of multi-organ involvement has been reported in SMA animal models and/or patients [17][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35].
Over the last few years, the increased life expectancy of SMA patients has been achieved through the invention of novel therapies and the standardization of clinical care. However, knowledge of the altered metabolism and nutrition in SMA remains limited. The impact of SMN deficiency on metabolic abnormalities has been recently proposed. Before and after the onset of the disease, metabolomics studies in SMA patients can provide valuable information about the direct or indirect effects of SMN deficiency on metabolic abnormalities [13]. The present review will discuss the current knowledge regarding the metabolic involvement in SMA and the role of metabolomics in pursuing potential biomarkers and therapeutic insights for SMA.
2. Lipid Metabolic Abnormalities in SMA
Abnormalities of lipid metabolism have been described in different motor neuron diseases, including SMA [36]. Dysregulated lipid metabolism is the first and most studied nutritional problem in SMA [37][38]. Compared with healthy controls and non-SMA motor neuron diseases with equally debilitating statuses, the abnormal lipid metabolism found in patients and animal models appears unique to SMA [39][40]. Abnormal levels of fatty acid oxidation metabolites, especially dicarboxylic aciduria and esterified carnitine, were first reported in several studies of patients with severe SMA type [38][41][42] Subsequently, an increasing number of studies suggest that patients with SMA are likely to have metabolic defects involving fatty acid metabolism. Of note, increased fat mass, even though relatively low caloric consumption has been repeatedly reported in patients with SMA [40][43]. Several serum fatty acids and lipids have been found correlated to the motor function of patients with SMA, suggesting potential biomarker candidates for SMA [44]. It has recently been implicated that defects in fatty acid transport and mitochondrial β-oxidation may also contribute to muscle wasting in patients with a severe SMA phenotype [32]. Nevertheless, the exact mechanism of this lipid metabolism abnormality in SMA is still unclear, but it is suspected to be related to the absence of the SMN gene product, defects in neighboring genes, or the loss of a neural “trophic factor” [31][42][45].
Although abnormal levels of fatty acid metabolites have been reported, no direct evidence has substantiated a specific defect of mitochondrial β-oxidation in SMA patients. There are several differences in metabolomics between patients with SMA and patients with a genetic defect of fatty acid β-oxidation. SMA patients usually had a normal acylcarnitine profile [42], contrary to an increased acylcarnitine level always found in mitochondrial β-oxidation defects. Moreover, fasting patients with impaired fatty acid β-oxidation always have markedly decreased ketone bodies. However, patients with SMA usually present with a normal or even a high ketone body level (increased ketosis), especially under stress [45][46]. The ability to mount fasting ketosis means that the liver can utilize fatty acids normally, but it does not rule out that it may be caused by muscle-specific mitochondrial defects in β-oxidation [32]. Therefore, it is postulated that dysregulated fatty acid metabolism in SMA patients might be directly related to SMN deficiency but is not attributed to the consequence of major enzyme block of mitochondrial β-oxidation, disuse muscle atrophy, or denervation [13][42][47].
Fatty vacuolization with macro- or micro-vesicular steatosis of the liver has been found in early studies of SMA patients [38][41][42]. Of note, liver failure and Reye-like syndrome with diffuse vesicular steatosis have been recently reported in patients with type 1 or 2 SMA [48][49]. An updated study further reports an increased susceptibility to develop dyslipidemia in 37% of SMA patients, with evidence of liver steatosis in their pathological specimens [27]. Similarly, these human findings are reproduced in different SMA mouse models, of which a specific Smn 2B/− mice model developed the non-alcoholic fatty liver disease (NAFLD) before denervation. Hyperglucagonemia might also contribute to dyslipidemia and hepatic steatosis, possibly through the pancreas–liver axis, leading to peripheral lipolysis of white adipose tissue and an increase in circulating lipids. These findings imply that the liver-intrinsic SMN deficiency might also cause dysregulated metabolism of the hepatocytes [26][50], which could predispose the cells to fat accumulation. Noteworthily, subacute liver failure was recently reported in two patients with type 1 SMA following gene replacement therapy [49]. It is postulated that increased susceptibility to dyslipidemia and associated fatty liver disease could predispose the SMA patient to liver injury, which might be induced or exacerbated after the gene therapy. A thorough investigation of the lipid content in the liver of SMA patients and mouse models, before and after the onset of the disease [47], may provide further evidence for whether the direct or indirect effects of SMN deficiency affect metabolic abnormalities.
Since carnitine and its acyl esters (acylcarnitines) are cofactors for β-oxidation, abnormal lipid metabolism may also be reflected in their production, fractions, and transportation. Because carnitine is essential for intramitochondrial β-oxidation, reduced carnitine would limit β-oxidation. Acylcarnitines are known to play a crucial role in stabilizing neuronal membranes and neurotransmission [51]. Supplementation of acylcarnitine has shown beneficial effects in treating chronic degenerative diseases [52][53]. However, there are still controversies regarding the dysregulation of production, synthesis, and carnitine/acylcarnitine extraction in SMA patients. Early studies suggested that the integrity of nerve and motor neurons might influence carnitine transportation and lipid β-oxidation in muscles. Reduced muscle carnitine and decreased activity of β-oxidation have been observed in animal models after denervation [54][55]. Similarly, reduced carnitine and acylcarnitine levels in plasma and muscles and increased urine excretion of acylcarnitine have been reported in SMA patients [37][56]. However, normal or mild-to-moderate elevated serum acylcarnitines, particularly C5-OH acylcarnitine and C3 propionylcarnitine, were found in the following studies of SMA patients with a severe phenotype [41][42]. In contrast, an updated article reported an adolescent with type 2 SMA who showed a dramatically low serum carnitine/acylcarnitine level at a catabolic state [48]. This finding suggests impaired intramitochondrial β-oxidation associated with dysregulated carnitine metabolism in SMA would become more prominent, especially under stress.
In the fat metabolism of healthy individuals, longer-chain fatty acids are transported into the mitochondria for β-oxidation. Carnitine palmitoyltransferase 1 (CPT1) is an enzyme that converts long-chain acyl-CoA into long-chain acylcarnitine, thereby transporting long-chain fatty acids to the mitochondria. Decreased CPT activity has been reported in muscles of severe type 1 SMA patients, compared with aged-matched infants [56]. Recently, reduced CPT1 activity was also found in an SMA (Smn 2B/−) mice model [25]. Of note, an isoform of CPT1, called CPT1c, which mainly expresses in neurons, including motor neurons, shows biosynthetic activity in neuron-specific acyl-CoA. Reduced activity of CPT1c leads to motor function impairment and muscle weakness [57]. Interestingly, an updated study indicates that MN-specific CPT1C can interact with atlastin-1 encoded by the ATL1 gene, which is mutated in hereditary spastic paraplegia, a kind of motor neuron degenerative disorder [58].
Acylcarnitines can also interact with different proteins to influence signaling pathways of neuronal cells [52]. Growth-associated protein 43 (GAP43), a protein involved in neuronal development, neurotransmission, and neuroplasticity, is modified post-translationally by a long-chain acylcarnitine, possibly through the acylation pathway [59]. Interestingly, a recent study found that motor neurons from SMA mouse models showed reduced GAP43 protein levels in axons and growth cones [60][61]. SMN seems to be responsible for regulating the localization and translation of GAP43 mRNA in these axons, and the restoration of GAP43 mRNA and protein levels rescued the defect of axon growth in SMA mice. Therefore, dysregulated acylcarnitine might also affect SMA phenotypes, possibly through the post-translational regulation of motor neuron-specific protein GAP43. Acylcarnitine plays a role in GAP43-related axon growth/repair pathways and may represent a promising SMA treatment target.
Nevertheless, the inconsistent findings of carnitine/acylcarnitine metabolites in SMA patients argue the pathomechanism of the impaired β-oxidation in SMA. Applying modern techniques for quantitative analysis of carnitine and acylcarnitine of various lengths in different samples (e.g., plasma, urine, and muscle) may help decipher this ambiguity [62][63]. However, similar studies in SMA patients are scarce, and the findings of changed carnitine/acylcarnitine levels in SMA patients with different SMN2 copies have not been updated. The discovery of plasma and urinary metabolite patterns, specifically reflective of fatty acid catabolism, can help clarify biochemical pathways that link lipid metabolism and provide potential biomarkers monitoring disease progression.
3. Dietary Issues in SMA
Patients with SMA are at higher risk of suboptimal nutrition intake, and nearly half of the cohort demonstrated either undernutrition (underweight) or overnutrition (overweight) over time [18][64]. Changes in body composition, especially the loss of lean body mass, can be particularly harmful to SMA patients because it can impair the respiratory strength of already weak muscles [43]. Therefore, nutrition support is considered a core component of multidisciplinary care for SMA patients [15][65].
However, the specific nutritional challenges in this population are not well described, and a particular diet has not been scientifically evaluated. An early study showed that when the mother was fed a lipid-rich diet, the pups of SMA mice could have a longer survival period and improved motor function [39]. These findings suggested that higher fat content may confer protective benefits during motor neuron loss. However, an updated study reported that low-fat diets could nearly double survival in Smn 2B/− mice, independent of changes in SMN levels, liver steatosis, or enhanced hepatic functions [66]. Although both studies are in the preclinical phase, such controversies suggest a need to establish clinical nutrition guidance from evidence-based research to provide better care for SMA patients.
The advances in therapy for SMA have improved survival and quality of life, which poses new challenges. The survival of patients with severe SMA has generated new phenotypes, and long-term outcomes are unknown [67]. Noteworthily, nutritional management may have a significant impact on the clinical course and even prognosis. For example, previous studies indicated that nutritional support could affect the therapeutic effects of trial agents on different SMA mice models [68][69]. Although it is difficult to show a clear association between metabolic effects in SMA patients who received therapies at this time, it has been emphasized that nutritional care must also be revised and monitored according to individual needs, especially in the SMA therapeutic era [15]. Optimal nutritional management for patients with SMA includes longitudinal evaluation of weight and length and dietary analysis. Recent studies have demonstrated that a modified diet based on measured energy expenditure and optimized protein can improve ventilation and lean body mass in patients with SMA [18][70]. In the future, non-invasive approaches for body composition assessment, e.g., bioelectrical impedance analysis, can be used to evaluate the nutritional status of children with SMA. Further research is needed to assess the use of elemental and semi-elemental formulas in SMA management, including the optimal intake of macronutrients and micronutrients for nutritional support and the ideal fat content and composition.
This entry is adapted from the peer-reviewed paper 10.3390/nu12123842
References
- Dubowitz, V. Ramblings in the history of spinal muscular atrophy. Neuromuscul. Disord. 2009, 19, 69–73.
- Darras, B.T. Spinal muscular atrophies. Pediatric Clin. N. Am. 2015, 62, 743–766.
- Lunn, M.R.; Wang, C.H. Spinal muscular atrophy. Lancet 2008, 371, 2120–2133.
- Farrar, M.A.; Park, S.B.; Vucic, S.; Carey, K.A.; Turner, B.J.; Gillingwater, T.H.; Swoboda, K.J.; Kiernan, M.C. Emerging therapies and challenges in spinal muscular atrophy. Ann. Neurol. 2017, 81, 355–368.
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165.
- Burghes, A.H. When is a deletion not a deletion? When it is converted. Am. J. Hum. Genet. 1997, 61, 9–15.
- Burghes, A.H.; Beattie, C.E. Spinal muscular atrophy: Why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009, 10, 597–609.
- Tisdale, S.; Pellizzoni, L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J. Neurosci. 2015, 35, 8691–8700.
- Wirth, B.; Karakaya, M.; Kye, M.J.; Mendoza-Ferreira, N. Twenty-five years of spinal muscular atrophy research: From phenotype to genotype to therapy, and what comes next. Annu. Rev. Genom. Hum. Genet. 2020, 21, 231–261.
- Tu, W.Y.; Simpson, J.E.; Highley, J.R.; Heath, P.R. Spinal muscular atrophy: Factors that modulate motor neurone vulnerability. Neurobiol. Dis. 2017, 102, 11–20.
- Chen, T.H. New and developing therapies in spinal muscular atrophy: From genotype to phenotype to treatment and where do we stand? Int. J. Mol. Sci. 2020, 21, 3297.
- Nash, L.A.; Burns, J.K.; Chardon, J.W.; Kothary, R.; Parks, R.J. Spinal muscular atrophy: More than a disease of motor neurons? Curr. Mol. Med. 2016, 16, 779–792.
- Simone, C.; Ramirez, A.; Bucchia, M.; Rinchetti, P.; Rideout, H.; Papadimitriou, D.; Re, D.B.; Corti, S. Is spinal muscular atrophy a disease of the motor neurons only: Pathogenesis and therapeutic implications? Cell Mol. Life Sci. 2016, 73, 1003–1020.
- Yeo, C.J.J.; Darras, B.T. Overturning the paradigm of spinal muscular atrophy as just a motor neuron disease. Pediatr. Neurol. 2020, 109, 12–19.
- Mercuri, E.; Finkel, R.S.; Muntoni, F.; Wirth, B.; Montes, J.; Main, M.; Mazzone, E.S.; Vitale, M.; Snyder, B.; Quijano-Roy, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul. Disord. 2018, 28, 103–115.
- Davis, R.H.; Miller, E.A.; Zhang, R.Z.; Swoboda, K.J. Responses to fasting and glucose loading in a cohort of well children with spinal muscular atrophy type ii. J. Pediatrics 2015, 167, 1362–1368.
- Bowerman, M.; Swoboda, K.J.; Michalski, J.P.; Wang, G.S.; Reeks, C.; Beauvais, A.; Murphy, K.; Woulfe, J.; Screaton, R.A.; Scott, F.W.; et al. Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann. Neurol. 2012, 72, 256–268.
- Mehta, N.M.; Newman, H.; Tarrant, S.; Graham, R.J. Nutritional status and nutrient intake challenges in children with spinal muscular atrophy. Pediatric Neurol. 2016, 57, 80–83.
- Lipnick, S.L.; Agniel, D.M.; Aggarwal, R.; Makhortova, N.R.; Finlayson, S.G.; Brocato, A.; Palmer, N.; Darras, B.T.; Kohane, I.; Rubin, L.L. Systemic nature of spinal muscular atrophy revealed by studying insurance claims. PLoS ONE 2019, 14, e0213680.
- Wishart, T.M.; Huang, J.P.; Murray, L.M.; Lamont, D.J.; Mutsaers, C.A.; Ross, J.; Geldsetzer, P.; Ansorge, O.; Talbot, K.; Parson, S.H.; et al. Smn deficiency disrupts brain development in a mouse model of severe spinal muscular atrophy. Hum. Mol. Genet. 2010, 19, 4216–4228.
- Mendonca, R.H.; Rocha, A.J.; Lozano-Arango, A.; Diaz, A.B.; Castiglioni, C.; Silva, A.M.S.; Reed, U.C.; Kulikowski, L.; Paramonov, I.; Cusco, I.; et al. Severe brain involvement in 5q spinal muscular atrophy type 0. Ann. Neurol. 2019, 86, 458–462.
- Somers, E.; Lees, R.D.; Hoban, K.; Sleigh, J.N.; Zhou, H.; Muntoni, F.; Talbot, K.; Gillingwater, T.H.; Parson, S.H. Vascular defects and spinal cord hypoxia in spinal muscular atrophy. Ann. Neurol. 2016, 79, 217–230.
- Wijngaarde, C.A.; Blank, A.C.; Stam, M.; Wadman, R.I.; van den Berg, L.H.; van der Pol, W.L. Cardiac pathology in spinal muscular atrophy: A systematic review. Orphanet J. Rare Dis. 2017, 12, 67.
- Schreml, J.; Riessland, M.; Paterno, M.; Garbes, L.; Rossbach, K.; Ackermann, B.; Kramer, J.; Somers, E.; Parson, S.H.; Heller, R.; et al. Severe sma mice show organ impairment that cannot be rescued by therapy with the hdaci jnj-26481585. Eur. J. Hum. Genet. 2013, 21, 643–652.
- Deguise, M.-O.; Pileggi, C.; Beauvais, A.; Tierney, A.; Chehade, L.; De Repentigny, Y.; Michaud, J.; Llavero-Hurtado, M.; Lamont, D.; Atrih, A.J.B. A mouse model for spinal muscular atrophy provides insights into non-alcoholic fatty liver disease pathogenesis. bioRxiv 2020.
- Szunyogova, E.; Zhou, H.; Maxwell, G.K.; Powis, R.A.; Muntoni, F.; Gillingwater, T.H.; Parson, S.H. Survival motor neuron (smn) protein is required for normal mouse liver development. Sci. Rep. 2016, 6, 34635.
- Deguise, M.O.; Baranello, G.; Mastella, C.; Beauvais, A.; Michaud, J.; Leone, A.; De Amicis, R.; Battezzati, A.; Dunham, C.; Selby, K.; et al. Abnormal fatty acid metabolism is a core component of spinal muscular atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 1519–1532.
- Nery, F.C.; Siranosian, J.J.; Rosales, I.; Deguise, M.O.; Sharma, A.; Muhtaseb, A.W.; Nwe, P.; Johnstone, A.J.; Zhang, R.; Fatouraei, M.; et al. Impaired kidney structure and function in spinal muscular atrophy. Neurol. Genet. 2019, 5, e353.
- Allardyce, H.; Kuhn, D.; Hernandez-Gerez, E.; Hensel, N.; Huang, Y.T.; Faller, K.; Gillingwater, T.H.; Quondamatteo, F.; Claus, P.; Parson, S.H. Renal pathology in a mouse model of severe spinal muscular atrophy is associated with downregulation of glial cell-line derived neurotrophic factor (gdnf). Hum. Mol. Genet. 2020, 29, 2365–2378.
- Sintusek, P.; Catapano, F.; Angkathunkayul, N.; Marrosu, E.; Parson, S.H.; Morgan, J.E.; Muntoni, F.; Zhou, H. Histopathological defects in intestine in severe spinal muscular atrophy mice are improved by systemic antisense oligonucleotide treatment. PLoS ONE 2016, 11, e0155032.
- Davis, R.H.; Godshall, B.J.; Seffrood, E.; Marcus, M.; LaSalle, B.A.; Wong, B.; Schroth, M.K.; Swoboda, K.J. Nutritional practices at a glance: Spinal muscular atrophy type i nutrition survey findings. J. Child. Neurol. 2014, 29, 1467–1472.
- Ripolone, M.; Ronchi, D.; Violano, R.; Vallejo, D.; Fagiolari, G.; Barca, E.; Lucchini, V.; Colombo, I.; Villa, L.; Berardinelli, A.; et al. Impaired muscle mitochondrial biogenesis and myogenesis in spinal muscular atrophy. JAMA Neurol. 2015, 72, 666–675.
- Deguise, M.O.; Boyer, J.G.; McFall, E.R.; Yazdani, A.; De Repentigny, Y.; Kothary, R. Differential induction of muscle atrophy pathways in two mouse models of spinal muscular atrophy. Sci. Rep. 2016, 6, 28846.
- Vai, S.; Bianchi, M.L.; Moroni, I.; Mastella, C.; Broggi, F.; Morandi, L.; Arnoldi, M.T.; Bussolino, C.; Baranello, G. Bone and spinal muscular atrophy. Bone 2015, 79, 116–120.
- Shanmugarajan, S.; Tsuruga, E.; Swoboda, K.J.; Maria, B.L.; Ries, W.L.; Reddy, S.V. Bone loss in survival motor neuron (smn(−/−) smn2) genetic mouse model of spinal muscular atrophy. J. Pathol. 2009, 219, 52–60.
- Schmitt, F.; Hussain, G.; Dupuis, L.; Loeffler, J.P.; Henriques, A. A plural role for lipids in motor neuron diseases: Energy, signaling and structure. Front. Cell Neurosci. 2014, 8, 25.
- Harpey, J.P.; Charpentier, C.; Paturneau-Jouas, M.; Renault, F.; Romero, N.; Fardeau, M. Secondary metabolic defects in spinal muscular atrophy type ii. Lancet 1990, 336, 629–630.
- Kelley, R.I.; Sladky, J.T. Dicarboxylic aciduria in an infant with spinal muscular atrophy. Ann. Neurol. 1986, 20, 734–736.
- Butchbach, M.E.; Rose, F.F., Jr.; Rhoades, S.; Marston, J.; McCrone, J.T.; Sinnott, R.; Lorson, C.L. Effect of diet on the survival and phenotype of a mouse model for spinal muscular atrophy. Biochem. Biophys. Res. Commun. 2010, 391, 835–840.
- Sproule, D.M.; Montes, J.; Montgomery, M.; Battista, V.; Koenigsberger, D.; Shen, W.; Punyanitya, M.; De Vivo, D.C.; Kaufmann, P. Increased fat mass and high incidence of overweight despite low body mass index in patients with spinal muscular atrophy. Neuromuscul. Disord. Nmd 2009, 19, 391–396.
- Tein, I.; Sloane, A.E.; Donner, E.J.; Lehotay, D.C.; Millington, D.S.; Kelley, R.I. Fatty acid oxidation abnormalities in childhood-onset spinal muscular atrophy: Primary or secondary defect(s)? Pediatric Neurol. 1995, 12, 21–30.
- Crawford, T.O.; Sladky, J.T.; Hurko, O.; Besner-Johnston, A.; Kelley, R.I. Abnormal fatty acid metabolism in childhood spinal muscular atrophy. Ann. Neurol. 1999, 45, 337–343.
- Poruk, K.E.; Davis, R.H.; Smart, A.L.; Chisum, B.S.; Lasalle, B.A.; Chan, G.M.; Gill, G.; Reyna, S.P.; Swoboda, K.J. Observational study of caloric and nutrient intake, bone density, and body composition in infants and children with spinal muscular atrophy type i. Neuromuscul. Disord. 2012, 22, 966–973.
- Finkel, R.S.; Crawford, T.O.; Swoboda, K.J.; Kaufmann, P.; Juhasz, P.; Li, X.; Guo, Y.; Li, R.H.; Trachtenberg, F.; Forrest, S.J.; et al. Candidate proteins, metabolites and transcripts in the biomarkers for spinal muscular atrophy (bforsma) clinical study. PLoS ONE 2012, 7, e35462.
- Mulroy, E.; Gleeson, S.; Furlong, M.J. Stress-induced ketoacidosis in spinal muscular atrophy: An under-recognized complication. J. Neuromuscul. Dis. 2016, 3, 419–423.
- Lakkis, B.; El Chediak, A.; Hashash, J.G.; Koubar, S.H. Severe ketoacidosis in a patient with spinal muscular atrophy. Cen Case Rep. 2018, 7, 292–295.
- Shababi, M.; Lorson, C.L.; Rudnik-Schoneborn, S.S. Spinal muscular atrophy: A motor neuron disorder or a multi-organ disease? J. Anat. 2014, 224, 15–28.
- Zolkipli, Z.; Sherlock, M.; Biggar, W.D.; Taylor, G.; Hutchison, J.S.; Peliowski, A.; Alman, B.A.; Ling, S.C.; Tein, I. Abnormal fatty acid metabolism in spinal muscular atrophy may predispose to perioperative risks. Eur. J. Paediatr. Neurol. 2012, 16, 549–553.
- Feldman, A.G.; Parsons, J.A.; Dutmer, C.M.; Veerapandiyan, A.; Hafberg, E.; Maloney, N.; Mack, C.L. Subacute liver failure following gene replacement therapy for spinal muscular atrophy type 1. J. Pediatr. 2020, 225, 252–258.e1.
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral smn restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126.
- Ferreira, G.C.; McKenna, M.C. L-carnitine and acetyl-l-carnitine roles and neuroprotection in developing brain. Neurochem. Res. 2017, 42, 1661–1675.
- Jones, L.L.; McDonald, D.A.; Borum, P.R. Acylcarnitines: Role in brain. Prog. Lipid Res. 2010, 49, 61–75.
- Pennisi, M.; Lanza, G.; Cantone, M.; D’Amico, E.; Fisicaro, F.; Puglisi, V.; Vinciguerra, L.; Bella, R.; Vicari, E.; Malaguarnera, G. Acetyl-l-carnitine in dementia and other cognitive disorders: A critical update. Nutrients 2020, 12, 1389.
- Czyzewski, K.; Stern, L.Z.; Sadeh, M.; Bahl, J.J. Changes in muscle carnitine during regeneration. Exp. Neurol. 1984, 86, 73–80.
- Czyzewski, K.; Stern, L.Z.; Sadeh, M.; Bahl, J.J. Altered rat skeletal muscle carnitine with age and after denervation. Muscle Nerve 1985, 8, 34–37.
- Bresolin, N.; Freddo, L.; Tegazzin, V.; Bet, L.; Armani, M.; Angelini, C. Carnitine and acyltransferase in experimental neurogenic atrophies: Changes with treatment. J. Neurol. 1984, 231, 170–175.
- Carrasco, P.; Jacas, J.; Sahun, I.; Muley, H.; Ramirez, S.; Puisac, B.; Mezquita, P.; Pie, J.; Dierssen, M.; Casals, N. Carnitine palmitoyltransferase 1c deficiency causes motor impairment and hypoactivity. Behav. Brain Res. 2013, 256, 291–297.
- Rinaldi, C.; Schmidt, T.; Situ, A.J.; Johnson, J.O.; Lee, P.R.; Chen, K.L.; Bott, L.C.; Fado, R.; Harmison, G.H.; Parodi, S.; et al. Mutation in cpt1c associated with pure autosomal dominant spastic paraplegia. JAMA Neurol. 2015, 72, 561–570.
- Liang, X.; Lu, Y.; Neubert, T.A.; Resh, M.D. Mass spectrometric analysis of gap-43/neuromodulin reveals the presence of a variety of fatty acylated species. J. Biol. Chem. 2002, 277, 33032–33040.
- Fallini, C.; Donlin-Asp, P.G.; Rouanet, J.P.; Bassell, G.J.; Rossoll, W. Deficiency of the survival of motor neuron protein impairs mrna localization and local translation in the growth cone of motor neurons. J. Neurosci. 2016, 36, 3811–3820.
- Fuller, H.R.; Gillingwater, T.H.; Wishart, T.M. Commonality amid diversity: Multi-study proteomic identification of conserved disease mechanisms in spinal muscular atrophy. Neuromuscul Disord. 2016, 26, 560–569.
- Okun, J.G.; Kolker, S.; Schulze, A.; Kohlmuller, D.; Olgemoller, K.; Lindner, M.; Hoffmann, G.F.; Wanders, R.J.; Mayatepek, E. A method for quantitative acylcarnitine profiling in human skin fibroblasts using unlabelled palmitic acid: Diagnosis of fatty acid oxidation disorders and differentiation between biochemical phenotypes of mcad deficiency. Biochim. Biophys. Acta 2002, 1584, 91–98.
- Aguer, C.; McCoin, C.S.; Knotts, T.A.; Thrush, A.B.; Ono-Moore, K.; McPherson, R.; Dent, R.; Hwang, D.H.; Adams, S.H.; Harper, M.E. Acylcarnitines: Potential implications for skeletal muscle insulin resistance. FASEB J. 2015, 29, 336–345.
- Martinez, E.E.; Quinn, N.; Arouchon, K.; Anzaldi, R.; Tarrant, S.; Ma, N.S.; Griffin, J.; Darras, B.T.; Graham, R.J.; Mehta, N.M. Comprehensive nutritional and metabolic assessment in patients with spinal muscular atrophy: Opportunity for an individualized approach. Neuromuscul. Disord. 2018, 28, 512–519.
- Chen, Y.S.; Shih, H.H.; Chen, T.H.; Kuo, C.H.; Jong, Y.J. Prevalence and risk factors for feeding and swallowing difficulties in spinal muscular atrophy types ii and iii. J. Pediatrics 2012, 160, 447–451.
- Deguise, M.O.; Chehade, L.; Tierney, A.; Beauvais, A.; Kothary, R. Low fat diets increase survival of a mouse model of spinal muscular atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 2340–2346.
- Mercuri, E.; Pera, M.C.; Scoto, M.; Finkel, R.; Muntoni, F. Spinal muscular atrophy—Insights and challenges in the treatment era. Nat. Rev. Neurol. 2020, 16, 706–715.
- Butchbach, M.E.; Singh, J.; Gurney, M.E.; Burghes, A.H. The effect of diet on the protective action of d156844 observed in spinal muscular atrophy mice. Exp. Neurol. 2014, 256, 1–6.
- Narver, H.L.; Kong, L.; Burnett, B.G.; Choe, D.W.; Bosch-Marce, M.; Taye, A.A.; Eckhaus, M.A.; Sumner, C.J. Sustained improvement of spinal muscular atrophy mice treated with trichostatin a plus nutrition. Ann. Neurol. 2008, 64, 465–470.
- Bertoli, S.; De Amicis, R.; Mastella, C.; Pieri, G.; Giaquinto, E.; Battezzati, A.; Leone, A.; Baranello, G. Spinal muscular atrophy, types i and ii: What are the differences in body composition and resting energy expenditure? Clin. Nutr. 2017, 36, 1674–1680.