The RAS family consists of membrane-associated small GTPases which play essential roles in cell survival, proliferation, and differentiation. There are four RAS protein isoforms in humans: HRAS, NRAS, and two splice variants, KRAS4A and KRAS4B.
- KRAS
- colorectal cancer
- RAS signalling
- cancer stem cells
- RAS-driven metastasis
- CRC
- KRAS structure
- KRAS mutations
1. Introduction
RAS proteins comprise two regions, the G domain and the C terminal hypervariable region. G domain sequences are highly homologous, while the hypervariable regions (HVR) differ across the RAS isoforms [1]. The G domain is responsible for catalytic activity and effector binding, while the function of the HVR is mainly binding to the plasma membrane [2]. The HVR region, which is responsible for plasma membrane (PM) association, undergoes a series of posttranslational modifications (PTMs) required for it to bind the membrane. Membrane attachment is indispensable for cellular RAS function [3]. One of the PTMs required for membrane localisation is prenylation, which occurs on the CAAX sequence at the C terminal end of the protein (Figure 1). This modification results in farnesylation of the cysteine residue in the CAAX motif by farnesyltransferase (FTase), which promotes proteolytic cleavage of the last three amino acids (AAX) by RAS-converting enzyme 1 (RCE1) [4]. Next, a methyltransferase catalyses carboxyl methylation of the new C terminus at the cysteine residue remaining from the CAAX motif, and this reaction neutralises the negatively charged end of the polypeptide chain and increases its membrane affinity [4][5]. These reactions, together called CAAX processing, boost the membrane affinity of RAS; however, they alone are insufficient to support strong PM association. Palmitoylation of one or two cysteine residues in the HVR region can increase the membrane binding affinity [3] (Figure 1). One exception is the KRAS4B isoform, on which no palmitoylation occurs due to its polybasic region upstream of the CAAX motif. This special region consists of eight positively charged lysine residues that can form electrostatic bonds with the negatively charged phospholipid headgroups of the PM, and along with the prenylated C terminus, it is sufficient for membrane association [4][6]. Many attempts have been made to block posttranslational modification of the HVR, especially RAS prenylation, with varying levels of success. Currently, there are two class of drugs that are clinically approved and used for anticancer therapy via inhibition of protein prenylation. These drugs have proved to be ineffective against CRC [4]; however, there may still be hope since FGTI-2734, a recently developed peptide inhibitor, can disrupt KRAS membrane localisation in various human cancer cell lines, including the DLD1 colon cancer cell line [7].
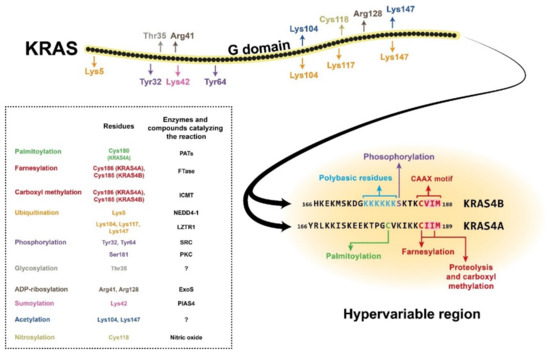
Figure 1. Structure of the KRAS4A and KRAS4B isoforms, showing the amino acid positions of posttranslational modifications with different colours. The hypervariable region sequences are presented separately for the two isoforms due to their high variability. The CAAX motifs and the polybasic residues on KRAS4B are also colour-coded. The posttranslational modifications are listed in the box with the affected amino acid sites and (where known) the enzyme or compound responsible for the reaction. NEDD4-1, neural precursor cell expressed developmentally downregulated 4-1; LZTR1, leucine-zipper-like transcriptional regulator 1; SRC, proto-oncogene tyrosine-protein kinase Src; PKC, protein kinase C; ExoS, exoenzyme S; PIAS4, protein inhibitor of activated STAT protein 4; PATs, protein acetyltransferases; FTase, farnesyltransferase; ICMT, isoprenylcysteine carboxyl methyltransferase.
In addition to prenylation and palmitoylation, many other PTMs can take place on RAS proteins (Figure 1). For example, phosphorylation of Ser181 of KRAS4B by PKC kinase can reduce the interaction of the protein with the PM via endocytic recycling [8]. The G domain has two other phosphorylation sites, Tyr32 and Tyr64, on which modifications can also downregulate RAS activity by inhibiting the binding affinity for its effectors [9]. There are other known posttranslational modifications on RAS proteins, e.g., polyubiquitination, which can target them for proteasomal degradation [10][11]. There are also many sites for mono- and polyubiquitination, but instead of downregulating RAS signalling, these modifications can enhance GTP binding and facilitate effector activation. Other PTMs, e.g., acetylation, sumoylation, ADP-ribosylation, glycosylation, and nitrosylation, are also assumed to influence KRAS regulation, but the exact functions are still poorly described [1][3] (Figure 1).
2. Mutant KRAS-Driven Enhanced Cell Signalling in CRC
Wild type KRAS functions as a controlled binary molecular switch, cycling between inactive and active signal-transducing conformations. Once KRAS is in its GTP-bound state, it undergoes structural changes that allow it to bind and cooperate with downstream signalling molecules. RAS signalling is prevented when GTP is hydrolysed and RAS is in its GDP-bound state. These two states are tightly regulated by GEFs (guanine nucleotide exchange factors) and GAPs (GTPase-activating proteins). GEFs fuel GDP release from RAS and facilitate GTP loading to activate RAS, while GAPs accelerate GTP hydrolysis, leading to RAS inactivation.
Essentially, the primary role of GTP-bound, active KRAS is to collect and activate multiple effector molecules at the membrane and to coordinate various signalling routes. In normal cells, upon ligand binding, receptor tyrosine kinases, e.g., EGFR, are autophosphorylated. Activated receptors then dimerise and recruit adaptor proteins (e.g., Grb2 or Shc) to their cytoplasmic tails. Via the adaptor proteins, GEFs are also associated with the molecular complex where they facilitate the conversion of inactive GDP-bound KRAS to the signal-transmitting, active GTP-bound form. Therefore, within KRAS molecules, the structure of the GTP/GDP interacting site determines its function.
The integrity of the GTP/GDP binding site in the KRAS G domain has special importance in maintaining the well-regulated function of KRAS, and a single amino acid change within this site can abolish normal regulation. Therefore, it is not surprising that CRC-associated mutations in KRAS are located within this effector site (Figure 2A). Single base substitutions in codons 12 and 13 are very common cancerogenic mutations that affect glycine residues in the GTP-binding pocket critical for GTPase function (Table 1). It is broadly accepted that these KRAS mutations lead to stabilisation of the protein in its prolonged active state, thereby amplifying the downstream signalling pathways.
The main and most robust KRAS-regulated signalling route is the MAPK pathway (Figure 2B). When KRAS is activated either traditionally via receptor activation or via oncogenic mutations, KRAS proteins dimerise. The KRAS dimers then bind and activate RAF kinases. Next, RAF can phosphorylate the two catalytic serine residues of MEK. MEK, a dual threonine and tyrosine recognition kinase, then phosphorylates other kinases in the pathway, namely ERK1 and ERK2. In normal cells, activated ERKs initiate multiple effector mechanisms, e.g., the G1/S-phase transition, inhibition of apoptosis, and cell motility [12]. Notably, in CRC cells, it has been demonstrated that KRAS mutations lead to aberrantly elevated MAPK signalling [13][14]. Therefore, many attempts have been made to target and inhibit molecules downstream of KRAS, although breakthrough success in therapeutic applications has not been realised [15].
Mutations in KRAS lead to increased proliferation of CRC cells, and in combination with other mutations, e.g., in the APC gene, promote tumorigenesis. Only in recent years has it been systematically described how concurrent misregulation of both MAPK and WNT signalling can lead to CRC. The key component of the crosstalk between the MAPK and WNT pathway is Glycogen synthase kinase 3β (GSK3β) [16]. GSK3β regulates proteasomal degradation of β-catenin and NRAS protein. GSK3β activation depends on APC and ERK activity; therefore, upon development of a loss-of-function mutation in APC, GSK3 no longer exerts its destabilising effect on β-catenin, and WNT signalling is enhanced [17]. When a KRAS mutation occurs along with an APC mutation during CRC development, mutant KRAS-driven MAPK signalling result in hyperphosphorylation of ERK, which further inhibits GSK3β function. Both malfunctioning APC and mutant KRAS-enhanced ERK activity synergistically inhibit GSK3-β to amplify the β-catenin effect [16]. Therefore, one possible therapeutic intervention against mutant KRAS would be to directly target GSK3β to increase its activation to deregulate β-catenin [18]. This type of trial has been recently performed by Lee et al., who treated CRC xenografts with small-molecule compounds to re-activate GSK3β-driven signalling [19].
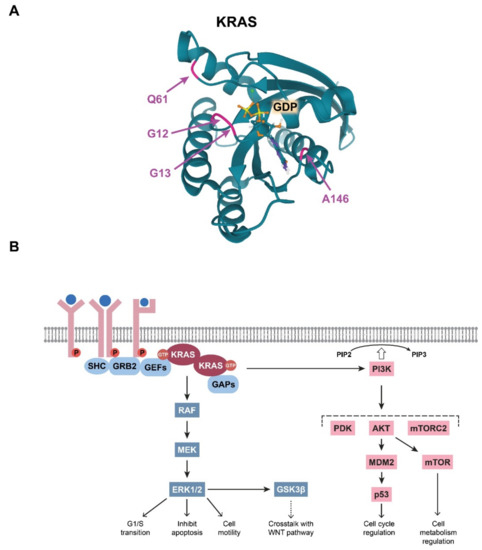
Figure 2. (A) Visualisation of the colorectal cancer (CRC)-related mutations in the KRAS G domain. The glutamine residue at position 61, the glycine residues at positions at 12 and 13, and the alanine residue at position 146, which form the GDP/GTP pocket, are indicated with colours. Image from the RCSB PDB (rcsb.org (accessed on 3 March 2021)) of PDB ID: 6MBT [20]. (B) Schematic representation of the summary of the RAS-driven signalling pathways.
3. Role of KRAS in CRC Stem Cells
Growing evidence suggests that a subset of cells within tumours, i.e., cancer stem cells (CSCs), possess stem cell properties such as self-renewal and differentiation drive heterogeneity within the tumour while also promoting cancer growth, proliferation, resistance, invasion, and metastasis [21]. This special cell type is, therefore, a promising target in the development of cancer therapies. To date, there are several hypotheses for how CRC develops [22]. According to the “top-down” hypothesis, more differentiated luminal cells dedifferentiate and regain stem cell properties, thereafter functioning as CSCs. The “bottom-up” hypothesis, however, argues that CRC originates from crypt base intestinal stem cells (ISCs). In both models, due to deregulated WNT signalling, initiating cells escape regulation and become CSCs [23]. Genetic alterations in both the WNT/β-catenin [19][24][25] and EGF/RAS [19][26][27][28] pathways drive CRC progression by influencing ISC self-renewal and proliferation, respectively, resulting in an abnormally sustained stem cell state. Initiating mutations, e.g., loss-of-function mutations in APC, cause robust WNT signalling deregulation [25], and when accompanied by mutations in other genes, e.g., KRAS (or components of TGFB, PI3 kinase, and TP53 signalling), can lead to malignant transformation [29][30].
KRAS is an important modulator of CSCs since its downstream effectors, the MAPK/ERK and PI3K/AKT pathways, are regulators of WNT/ β-catenin [29][31] signalling. However, an activating mutation in KRAS alone is insufficient to initiate CSCs [29][32]. As discussed above, APC mutations paired with oncogenic KRAS are activators of CSCs, driving malignant transformation and metastasis. The underlying mechanism was shown to be that APC loss leads to both nuclear accumulation of β-catenin and stabilisation of oncogenic KRAS which, then, via the ERK pathway, further enhances and fuels activated WNT/β-catenin signalling and establishment of CSC characteristics, including enhanced sphere-forming capacity, tumour size and weight, and expression of CSC markers (e.g., CD44, CD133, and CD166) [29][33]. The importance of KRAS stability in CSC activation was also demonstrated by work on WD repeat protein 76 (WDR76), an E3 ligase that destabilises RAS, thereby acting as a tumour suppressor [34]. Ro et al. demonstrated that in APC-mutant CRC tumours, cytosolic WDR76 acts as a tumour suppressor by binding to and degrading KRAS via polyubiquitination-dependent proteasomal degradation, ultimately causing decreased activation of the WNT/β-catenin pathway. WDR76 deficiency increased oncogenic KRAS, ERK, AKT, and β-catenin levels and enhanced the expression of the CSC markers LGR5, CD44, CD133, and CD166 in APC-mutant DLD-1 CRC cells [34]. Hwang et al. identified REG4 as the most significant mediator (among other genes such as IL8, PHLDA1, S100A4, S100A6, and PROM1) in the promotion of CSC properties by oncogenic KRAS. REG4 was found to act through WNT/β-catenin signalling at the level of receptor activation in KRAS/APC double-mutant CRC cells [28].
In colon cells, LGR5, a WNT signalling component, initiates an intestinal stem cell (ISC) program; however, abnormal LGR5 expression does not initiate premalignant colon adenomas unless other genes, e.g., APC or CTNNB1 (β-catenin), are also mutated to drive aberrant WNT signalling. The progression from a premalignant state to malignant status can be fuelled by oncogenic KRAS [35]. Le Rolle et al. recently demonstrated an alternative de-differentiation model for colon adenoma progression into CRC. They compared gene expression in stage I colon carcinomas with that in benign colon adenomas in human colon tissue and showed that while LGR5 expression correlated with an ISC signature in wild type KRAS (stage I) colon carcinomas, the presence of oncogenic KRAS, regardless of LGR5 expression, was associated with an embryonic stem cell (ESC)-like transcriptional signature instead of an ISC program. This ability of oncogenic KRAS was demonstrated in mutant KRAS-transfected SW48 human colon cancer cells as well, where the induction of an ESC-like program was indicated by upregulation of factors required for reprogramming and ESC and colon cancer maintenance, e.g., SOX2, FGFR1, LCK, validated at both the mRNA and protein levels. Increased soft agar colony growth and correspondingly increased in vivo tumour growth were also observed. Such an ESC-like program is foreign to colon cells, representing a distinct route for CRC formation in the presence of oncogenic KRAS. miR145, an inhibitor of the embryonic stem cell program, was found to counteract the effects of mutant KRAS by suppressing malignant growth and promoting the differentiation of mutant KRAS colon cancer cells [36].
As discussed above, oncogenic KRAS is a key driver of CRC progression from premalignant colon adenomas to stage I colon carcinomas, and it acts as an activator of CSCs in tumours. The expression levels of the CSC markers CD44 and CD166 correlate with higher risk of lymph node involvement and liver and lung metastasis in CRC patients carrying KRAS mutations [37]. Thus, it is of great importance to develop new strategies for targeting oncogenic KRAS and its related pathways in the context of CSC activation and maintenance.
This entry is adapted from the peer-reviewed paper 10.3390/cells10030667
References
- Wang, W.; Yuan, T.; Qian, M.; Yan, F.; Yang, L.; He, Q.; Yang, B.; Lu, J.; Zhu, H. Post-translational modification of KRAS: Potential targets for cancer therapy. Acta Pharmacol. Sin. 2020.
- Goswami, D.; Chen, D.; Yang, Y.; Gudla, P.; Columbus, J.; Worthy, K.; Rigby, M.; Wheeler, M.; Mukhopadhyay, S.; Powell, K.; et al. Membrane interactions of the globular domain and the hypervariable region of KRAS4b define its unique diffusion behavior. Elife 2020, 9.
- Ahearn, I.; Zhou, M.; Philips, M.R. Posttranslational Modifications of RAS Proteins. Cold Spring Harb. Perspect. Med. 2018, 8, a031484.
- Baranyi, M.; Buday, L.; Hegedűs, B. K-Ras prenylation as a potential anticancer target. Cancer Metastasis Rev. 2020, 39, 1127–1141.
- Hancock, J.F.; Cadwallader, K.; Marshall, C.J. Methylation and proteolysis are essential for efficient membrane binding of prenylated p21K-ras(B). EMBO J. 1991, 10, 641–646.
- Hancock, J.F.; Paterson, H.; Marshall, C.J. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 1990, 63, 133–139.
- Kazi, A.; Xiang, S.; Yang, H.; Chen, L.; Kennedy, P.; Ayaz, M.; Fletcher, S.; Cummings, C.; Lawrence, H.R.; Beato, F.; et al. Dual farnesyl and geranylgeranyl transferase inhibitor thwarts mutant KRAS-driven patient-derived pancreatic tumors. Clin. Cancer Res. 2019, 25, 5984–5996.
- Cho, K.; Casteel, D.E.; Prakash, P.; Tan, L.; van der Hoeven, D.; Salim, A.A.; Kim, C.; Capon, R.J.; Lacey, E.; Cunha, S.R.; et al. AMPK and Endothelial Nitric Oxide Synthase Signaling Regulates K-Ras Plasma Membrane Interactions via Cyclic GMP-Dependent Protein Kinase 2. Mol. Cell. Biol. 2016, 36, 3086–3099.
- Buday, L.; Vas, V. Novel regulation of Ras proteins by direct tyrosine phosphorylation and dephosphorylation. Cancer Metastasis Rev. 2020, 39, 1067–1073.
- Abe, T.; Umeki, I.; Kanno, S.I.; Inoue, S.I.; Niihori, T.; Aoki, Y. LZTR1 facilitates polyubiquitination and degradation of RAS-GTPases. Cell Death Differ. 2020, 27, 1023–1035.
- Zeng, T.; Wang, Q.; Fu, J.; Lin, Q.; Bi, J.; Ding, W.; Qiao, Y.; Zhang, S.; Zhao, W.; Lin, H.; et al. Impeded Nedd4-1-Mediated Ras Degradation Underlies Ras-Driven Tumorigenesis. Cell Rep. 2014, 7, 871–882.
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK signalling: A master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632.
- Benvenuti, S.; Sartore-Bianchi, A.; Di Nicolantonio, F.; Zanon, C.; Moroni, M.; Veronese, S.; Siena, S.; Bardelli, A. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007, 67, 2643–2648.
- Korzeniecki, C.; Priefer, R. Targeting KRAS mutant cancers by preventing signaling transduction in the MAPK pathway. Eur. J. Med. Chem. 2020, 113006.
- Yuan, X.; Tang, Z.; Du, R.; Yao, Z.; Cheung, S.H.; Zhang, X.; Wei, J.; Zhao, Y.; Du, Y.; Liu, Y.; et al. RAF dimer inhibition enhances the antitumor activity of MEK inhibitors in K-RAS mutant tumors. Mol. Oncol. 2020, 14, 1833–1849.
- Lee, S.K.; Hwang, J.H.; Choi, K.Y. Interaction of the Wnt/β-catenin and RAS-ERK pathways involving co-stabilization of both β-catenin and RAS plays important roles in the colorectal tumorigenesis. Adv. Biol. Regul. 2018, 68, 46–54.
- Jeong, W.-J.; Ro, E.J.; Choi, K.-Y. Interaction between Wnt/β-catenin and RAS-ERK pathways and an anti-cancer strategy via degradations of β-catenin and RAS by targeting the Wnt/β-catenin pathway. NPJ Precis. Oncol. 2018, 2.
- Lee, S.; Jeong, W.; Cho, Y.; Cha, P.; Yoon, J.; Ro, E.J.; Choi, S.; Oh, J.; Heo, Y.; Kim, H.; et al. β-Catenin-RAS interaction serves as a molecular switch for RAS degradation via GSK 3β. EMBO Rep. 2018, 19.
- Lee, S.-K.; Cho, Y.-H.; Cha, P.-H.; Yoon, J.-S.; Ro, E.J.; Jeong, W.-J.; Park, J.; Kim, H.; Kim, T.I.; Min, D.S.; et al. A small molecule approach to degrade RAS with EGFR repression is a potential therapy for KRAS mutation-driven colorectal cancer resistance to cetuximab. Exp. Mol. Med. 2018.
- Dharmaiah, S.; Tran, T.H.; Messing, S.; Agamasu, C.; Gillette, W.K.; Yan, W.; Waybright, T.; Alexander, P.; Esposito, D.; Nissley, D.V.; et al. Structures of N-terminally processed KRAS provide insight into the role of N-acetylation. Sci. Rep. 2019, 9, 10512.
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018.
- Huels, D.J.; Sansom, O.J. Stem vs non-stem cell origin of colorectal cancer. Br. J. Cancer 2015.
- Basu, S.; Haase, G.; Ben-Ze’ev, A. Wnt signaling in cancer stem cells and colon cancer metastasis. F1000Research 2016.
- Zhou, Y.; Xia, L.; Wang, H.; Oyang, L.; Su, M.; Liu, Q.; Lin, J.; Tan, S.; Tian, Y.; Liao, Q.; et al. Cancer stem cells in progression of colorectal cancer. Oncotarget 2018, 9.
- Novellasdemunt, L.; Antas, P.; Li, V.S.W. Targeting Wnt signaling in colorectal cancer. A Review in the Theme: Cell Signaling: Proteins, Pathways and Mechanisms. Am. J. Physiol. Physiol. 2015, 309, C511–C521.
- Feng, Y.; Dai, X.; Li, X.; Wang, H.; Liu, J.; Zhang, J.; Du, Y.; Xia, L. EGF signalling pathway regulates colon cancer stem cell proliferation and apoptosis. Cell Prolif. 2012.
- Koveitypour, Z.; Panahi, F.; Vakilian, M.; Peymani, M.; Seyed Forootan, F.; Nasr Esfahani, M.H.; Ghaedi, K. Signaling pathways involved in colorectal cancer progression. Cell Biosci. 2019, 9, 97.
- Hwang, J.; Yoon, J.; Cho, Y.; Cha, P.; Park, J.; Choi, K. A mutant KRAS-induced factor REG4 promotes cancer stem cell properties via Wnt/β-catenin signaling. Int. J. Cancer 2020, 146, 2877–2890.
- Moon, B.-S.; Jeong, W.-J.; Park, J.; Kim, I.; Min, S.; Choi, K.-Y. Role of Oncogenic K-ras in cancer Stem cell Activation by Aberrant Wnt/β-catenin Signaling. Oxf. J. 2014, 106.
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767.
- Fearon, E.R.; Wicha, M.S. KRAS and cancer stem cells in APC-mutant colorectal cancer. J. Natl. Cancer Inst. 2014, 106.
- Sansom, O.J.; Meniel, V.; Wilkins, J.A.; Cole, A.M.; Oien, K.A.; Marsh, V.; Jamieson, T.J.; Guerra, C.; Ashton, G.H.; Barbacid, M.; et al. Loss of Apc allows phenotypic manifestation of the transforming properties of an endogenous K-ras oncogene in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 14122–14127.
- Jeong, W.J.; Yoon, J.; Park, J.C.; Lee, S.H.; Lee, S.H.; Kaduwal, S.; Kim, H.; Yoon, J.B.; Choi, K.Y. Ras stabilization through aberrant activation of Wnt/β-catenin signaling promotes intestinal tumorigenesis. Sci. Signal. 2012, 5.
- Ro, E.J.; Cho, Y.H.; Jeong, W.J.; Park, J.C.; Min, D.S.; Choi, K.Y. WDR76 degrades RAS and suppresses cancer stem cell activation in colorectal cancer. Cell Commun. Signal. 2019, 17, 88.
- Phelps, R.A.; Chidester, S.; Dehghanizadeh, S.; Phelps, J.; Sandoval, I.T.; Rai, K.; Broadbent, T.; Sarkar, S.; Burt, R.W.; Jones, D.A. A Two-Step Model for Colon Adenoma Initiation and Progression Caused by APC Loss. Cell 2009, 137, 623–634.
- Le Rolle, A.F.; Chiu, T.K.; Zeng, Z.; Shia, J.; Weiser, M.R.; Paty, P.B.; Chiu, V.K. Oncogenic KRAS activates an embryonic stem cell-like program in human colon cancer initiation. Oncotarget 2016, 7, 2159–2174.
- Ribeiro, K.B.; Da Silva Zanetti, J.; Ribeiro-Silva, A.; Rapatoni, L.; De Oliveira, H.F.; Da Cunha Tirapelli, D.P.; Garcia, S.B.; Feres, O.; Da Rocha, J.J.R.; Peria, F.M. KRAS mutation associated with CD44/CD166 immunoexpression as predictors of worse outcome in metastatic colon cancer. Cancer Biomark. 2016, 16, 513–521.