Uveal melanoma (UM), the most common intraocular malignancy in adults, is a rare subset of melanoma. Despite effective primary therapy, around 50% of patients will develop the metastatic disease. Several clinical trials have been evaluated for patients with advanced UM, though outcomes remain dismal due to the lack of efficient therapies. Epigenetic dysregulation consisting of aberrant DNA methylation, histone modifications, and small non-coding RNA expression, silencing tumor suppressor genes, or activating oncogenes, have been shown to play a significant role in UM initiation and progression. Given that there is no evidence any approach improves results so far, adopting combination therapies, incorporating a new generation of epigenetic drugs targeting these alterations, may pave the way for novel promising therapeutic options. Furthermore, the fusion of effector enzymes with nuclease-deficient Cas9 (dCas9) in clustered regularly interspaced short palindromic repeats (CRISPR) associated protein 9 (Cas9) system equips a potent tool for locus-specific erasure or establishment of DNA methylation as well as histone modifications and, therefore, transcriptional regulation of specific genes.
- uveal melanoma
- epigenetic therapy
- DNA methylation
- histone modifications
- CRISPR-dCas9
- epigenetic editing
1. Biology and Molecular Subtypes of Uveal Melanoma
Uveal melanoma (UM) is the most common primary cancer of the eye, causing fatal liver metastasis in up to half of the patients [1][2]. The average annual incidence varies widely according to ethnicity or latitude between less than 1 to more than 9 per million population per year, with the highest incidence in white Caucasians [3][4]. Primary UM is treated with either surgery or radiation with a low local recurrence rate. However, there are no efficient therapies for metastatic UM, and as a result, most of the patients survive less than 12 months after metastases diagnosis [5]. A recent meta-analysis including 912 metastatic UM patients reported the median OS 10.2 months (95% CI 9.5–11.0) [6]. The UM tumors arise from melanocytes located in the uveal layer of the eye, with the choroid the most frequent site (82%), followed by the ciliary body (15%) and iris (3%) [7]. G protein subunit alpha q (GNAQ) and G protein subunit alpha 11 (GNA11) hotspot mutations, present in 83% of UM, are considered to be initiating events in UM tumorigenesis [8][9]. Recurrent cytogenetic abnormalities, including the most significant loss of one copy of chromosome 3 (M3) as well as 8p loss/8q gain, 6p gains, and 1p deletion, also hold prognostic potential [10]. Loss-of-function mutations in the BRCA1 associated protein 1 (BAP1) gene, located on 3p21, accompanied by decreased BAP1 mRNA and protein expression, have been identified in M3-UM, indicating that BAP1 abnormalities are highly correlated with the development of UM metastases. Patients with BAP1 mutations are generally younger, between 30 and 59 years, compared to the mean age at diagnosis, 62 years [11][12]. Gene expression profiling is considered an important prognostic tool that can predict metastatic risk with higher certainty than clinical stage or chromosome 3 status. A commercially available expression panel of 15 genes, established by Castle Biosciences, categorizes patients as Class 1 (low metastatic risk) or Class 2 (high metastatic risk) [13]. Class 1 patients are subdivided into Class 1a and Class 1b categories based on the key mutations and the number of 8q and 6p copies . The 5-year screening of patients revealed significant differences in metastatic risk of Class 1a, 1b, and Class 2 UMs, which are 2%, 21%, and 72%, respectively [14]. Recently, Robertson and colleagues identified four molecularly distinct UM subtypes, two associated with poor-prognosis M3 and two associated with better-prognosis disomy 3 (D3). Two subgroups of M3 tumors possess diverse clinical outcomes and are related to different biological pathways. In the first one, DNA damage repair, hypoxia-inducible factor 1 alpha (HIF1A), and MYC signaling are more prominent, while high levels of mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K)/Akt are more noticeable in the other subgroup [15]. This highlights the necessity for tailored therapeutic strategies that target these subtype-specific molecular changes. Moreover, unsupervised consensus clustering of the most variable 1% of CpG probes brought in four methylation clusters strongly associated with prognostic groups. UM in DNA methylation clusters 2 and 3 were highly enriched (12 of 16 tumors) in SF3B1/SRFR2 mutations, whereas EIF1AX mutant tumors were only present in DNA methylation cluster 1. Thus, D3 UM with EIF1AX versus SF3B1/SRFR2 mutations conveys diverse DNA methylation patterns. M3 BAP1-aberrant UM tumors offered a single global DNA methylation profile [15].
2. Role of Epigenetic Changes in UM Progression
Epigenetic alterations can result in aberrant gene regulation, thereby playing an essential role in tumorigenesis. Therefore, a complete overview of the epigenomic landscape is required to track the molecular events involved in the initiation and progression of a particular disease. Epigenetic changes that, for example, silence tumor suppressor genes or activate oncogenes include DNA methylation, histone modifications, and small non-coding RNAs. Many of them have been associated with UM initiation and progression (Figure 1) [16].
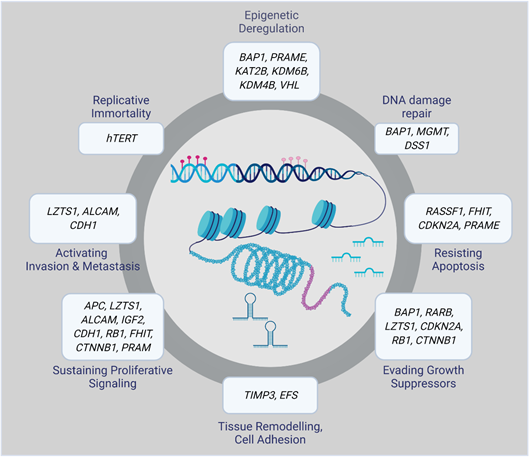
Figure 1. Epigenetically altered genes in uveal melanoma (UM).
2.1. DNA Methylation
DNA methylation is a covalent modification with the addition of a methyl group [-CH3] to the cytosine residue in the CpG dinucleotide sequence. Methylation/demethylation is an essential mechanism in maintaining cell- or tissue-specific gene expression[17] . Correlation between gene expression profiles with global DNA methylome clusters identified so far in prognostically distinct UM tumors, suggests an epigenetic contribution to the underlying molecular pathology that produces this transcriptome [18].
Preferentially Expressed Antigen in Melanoma (PRAME) is an emerging epigenetic biomarker of metastasis in low-risk UM tumors [19]. It was shown that hypomethylation of specific CpG sites nearby the PRAME promoter resulted in its transcriptional activation, correlated with high metastatic risk in both classes 1 UMs [20]. There was a significant association between the high expression level of the Deleted Split hand/Split foot 1 (DSS1) gene and some clinical-pathological features. A recent study discovered that 64% of UMs showed higher expression of DSS1 than healthy tissues. It was demonstrated that there is an inverse correlation between DSS1 expression activity and the methylation status of its promoter [21].
Aberrant promoter hypermethylation of CpG islands plays a critical role in the inactivation of tumor suppressor genes in cancer [22]. Hypermethylation of p16, TIMP3, RASSF1A, RASEF, hTERT, and EFS genes have been reported in UM [23][24][25][26][27][28].
The Ras association domain family 1 isoform A (RASSF1A) gene is located on chromosome 3p21.3, and its absence or inactivation has been proved to be a contributing factor in UM tumor formation and progression [29]. It plays a crucial role in cell-cycle regulation, apoptosis, and microtubule stability [30]. Methylation of promoter sites of this gene control entry at the retinoblastoma checkpoint and inhibits cyclin D1 protein accumulation at the post-transcriptional level, leading to cell-cycle progression block from the G1 to the S phase. Though the methylation of RASSF1A may not be wholly responsible for UM development, it could be a contributing factor in UM tumorigenesis . M3, which is related to the tumor’s metastatic capacity, has been reported in approximately half of all UMs. Considering the position of RASSF1A on the p21.3 region of chromosome 3, it could serve as a tumor suppressor gene whose silencing by methylation acts as a ‘second hit’ after monosomy occurs .
The Ras and EF-hand domain-containing (RASEF) gene, located on chromosome 9, region q21 is a candidate tumor suppressor gene. In 2007, UM cell lines and primary UM samples were screened for mutations in the RASEF gene region. The authors discovered that all cell lines and samples that did not express RASEF contained a methylated promoter, although those with RASEF expression lacked this methylation. They also demonstrated that methylation not only co-occur with low expression but also with a homozygous genotype. These findings propose that a combination of methylation and loss of heterozygosity may be the mechanism for loss of RASEF expression . Homozygous tumors with a methylated RASEF promoter region tend to display reduced survival compared with heterozygous tumors without methylation, suggesting loss of heterozygosity might be related to the aggressive behavior of the tumor [31].
A study by Venza and colleagues in 2015 showed that DNA methyltransferase 1 (Dnmt1) and Dnmt3b have a preeminent role in P16INK4A (alias CDKN2A) repression. They demonstrated that epigenetic alterations in the P16INK4A and P14ARF (the alternative reading frame protein product of the CDKN2A locus) genes were frequently associated with cutaneous as well as UMs [32]. Moreover, it was demonstrated that P16INK4A is frequently inactivated by hypermethylation in both primary UM and UM cell lines, accompanied by a down-regulated expression of P16INK4A . Both these studies also reported that loss of P16INK4A expression, attributable to CpG methylation, could be reversed when treated with the demethylating drug 5-aza-2’-deoxycytidine (decitabine). Interestingly, in UM patients who possess a tumor with a methylated P16INK4A promoter, metastasis tends to be more common. Therefore, modulation of abnormal methylation could be considered a valid target for UM treatment .
A recent study by Field and colleagues indicates that hypermethylation on chromosome 3 correlated with down-regulated gene expression at several loci, including 3p21 where BAP1 is located. All Class 2 tumors contained a novel hypermethylated site within the BAP1 locus, which reveals that BAP1 itself is epigenetically regulated. In functional validation experiments, Bap1 knockdown in UM cell lines consisted of a similar methylomic repatterning with UM tumors, enhanced for genes involved in axon guidance, melanogenesis, and development [33]. This study provides evidence that BAP1 loss leads to large-scale methylomic repatterning resulting in the Class 2 phenotype. Deciphering the role of epigenetic deregulation could explain the loss of melanocytic differentiation and gain of neural crest-like migratory behavior in Class 2 UMs.
2.2. Histone Modifications
Histone modification refers to the process of acetylation, phosphorylation, histone methylation, polyadenylation, ubiquitination, and ADP ribosylation, achieved by the relevant enzymatic activity [34]. Depletion of Bap1 protein trigger hyperubiquitination of H2A in melanoma cells and melanocytes, bringing about loss of differentiation along with the gain of stem-like properties[35][36] . On the contrary, treatment with histone deacetylase inhibitors (HDACis) in vivo in a xenograft model reversed the H2A hyperubiquitination, which may have therapeutic potential for inducing prolonged dormancy of micrometastatic UM disease [35]. While Hdac4 is localized to the nucleus in BAP1-mutant UM cells and the cells in which a BAP1 mutation was introduced using CRISPR-Cas9, it is restricted mainly to the cytoplasm in BAP1 wild-type UM cells and in normal human uveal melanocytes. Hence, Bap1 can inhibit the epigenetic function of Hdac4, at least in part, by diminishing its localization to the nucleus. Besides, short hairpin RNA (shRNA)-mediated depletion of Hdac4 in BAP1-mutant UM cells significantly impeded cell proliferation [37]. These findings suggest novel insights into the role of BAP1 in development and cancer and propose HDACis as potential therapeutic agents for BAP1-mutant cancer’s treatment.
2.3. miRNA-Based Epigenetic Mechanism
Micro RNAs (miRNA) are among the most studied non-coding RNAs. They are short (17–22 nucleotides in length), phylogenetically preserved single-stranded RNA molecules involved in the gene expression regulation. Their dysregulation has been ascertained to confer resistance to apoptosis, promote cell-cycle progression, and enhance invasiveness and metastasis of many cancers [38]. A significant number of miRNAs have been shown to be differentially expressed in UM cell lines and tumor tissues [39]. The expression level of let-7b, miR-143, miR-193b, miR-199a, and miR-652 were proved to be increased in Class 2 UMs, so they can be used to differentiate between Class 1 and Class 2 UM tumors [40]. Radhakrishnan and colleagues identified distinct miRNAs present in metastatic UM and absent in non-metastasizing tumors [41].
Moreover, 96 miRNAs were reported altered in UM cell lines. Among them, 65 were downregulated, 28 upregulated, and 3 exhibited a different expression pattern [42]. The pleiotropic nature makes miRNAs particularly attractive drug targets. MiR-27a is an oncogenic miRNA overexpressed in various cancer types. Genistein was found to inhibit miR-27a expression in highly aggressive UM cells in vitro and in vivo, thus increasing the expression of its target gene ZBTB10 significantly. Therefore, the authors hypothesized that genistein growth inhibitory activities can be mediated via miR-27a regulatory mechanism [43].
Treatment of UM cells with a DNA hypomethylating agent decitabine and HDACi trichostatin A (TSA), can regulate miR-124a expression level via epigenetic mechanisms [44]. Furthermore, decitabine was capable of enhancing miR-137 expression, which is generally epigenetically silenced during UM initiation [45], proving that individual epigenetic mechanisms can interact with each other. The list of UM-associated miRNAs (reviewed in [46][47]) is continually expanding along with the development of experimental methods and miRNA research tools. However, the identification of clinically relevant target genes and corresponding biological pathways will pave the way for their therapeutic targeting.
3. Epigenetic Editing
Due to the reversible nature of epigenetic changes, converting them to a “normal-like” chromatin landscape is feasible using “designed modular tools” [48]. The fundamental structure of editing tools includes a programmable DNA-binding domain and an epigenetic effector domain of interest. The zinc finger nucleases (ZFN), transcription activator-like effector (TALE), and nuclease-deficient Cas9 (dCas9) are three different programmable DNA-recognition domains which have been broadly used in epigenome editing, the most promising derivative technology of genome editing [49]. The epigenetic effectors are diverse, ranging from the writers, erasers, and readers, depositing, removing, or detecting DNA and histone modification marks as well as artificial transcription factors [50].
3.1. Epigenetic Editing Using CRISPR/dCas9
The clustered regularly interspaced short palindromic repeats (CRISPR) bacteria primitively used associated protein 9 (Cas9) system for their defense against bacteriophages. Recently this system is receiving remarkable attention by its rising role in the treatment of genetic disorders and cancer [51]. CRISPR system consists of an RNA-guided Cas9 endonuclease protein, which has the potential to be repurposed for editing both the genome and epigenome with significant efficiency [52]. This system can dramatically accelerate cancer research progress, either by screening for novel therapeutic targets, developing cancer therapies or by functional genome/epigenome editing. Therefore, it can be considered a robust weapon in the arsenal of future cancer treatment [51][52].
Two de novo DNA methyltransferases (Dnmt3a/b) are responsible for establishing DNA methylation in mammalian cells and Dnmt1 for its maintaining. Furthermore, demethylation is feasible through oxidation of the methyl group by TET (ten–eleven translocation) dioxygenases to build 5-hydroxymethylcytosine, and then its recovery into unmodified cytosine by whether DNA glycosylase-initiated base excision repair or DNA replication-dependent dilution [53].
It was demonstrated that fusion of mutant form of Cas9 without endonuclease activity known as dCas9, with the Dnmt3a or Tet1 enzyme could provide a potent tool for targeted erasure or establishment of DNA methylation, respectively. In 2016 Rudolf Jaenisch group successfully repurposed the CRISPR/Cas9 system to rearrange the targeted genomic sequences’ methylation status. dCas9 protein was fused either to the Dnmt3a (dCas9-Dnmt3a) or the catalytic domain of Tet1 (dCas9-Tet1) to predictably edit the epigenetic state of specific sequences [54].
To methylate genomic sites of interest by raising the local Dnmt3a concentration dCas9 protein was fused to repetitive peptide epitopes (SunTag), recruiting multiple copies of antibody-fused Dnmt3a (dCas9-SunTag-Dnmt3a). The authors reported that dCas9-SunTag-Dnmt3a can significantly boost CpG methylation at the HOXA5 locus in human embryonic kidney (HEK293T) cells and thus inhibit HOXA5 gene expression, not to mention its minimal impact on the global DNA methylome and transcriptome [55].
By gaining insight into the crucial role of molecular abnormalities, including epigenetic changes in cancer initiation, progression, and metastasis, there is a great demand for modern technologies to restore these aberrations. Therefore, in the coming years, the CRISPR-Cas system will play a notable role in cancer research. Repurposing this system to modulate the epigenome of cancer cells grant novel cancer therapies, albeit the employment of this platform, poses enormous challenges . Taking together, dCas9-Dnmt3a/Tet1 could be an appropriate tool for implementing the new therapeutic strategies for UM (Figure 2) and other cancer types.
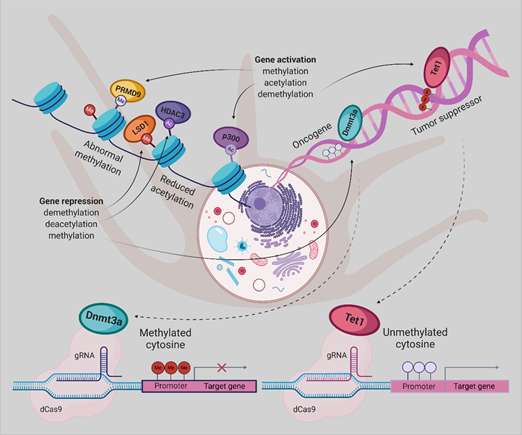
Figure 2. Epigenetic editing by CRISPR/dCas9, allowing for locus-specific control of epigenetically regulated gene expression, provides a more specific alternative to epigenetic drugs. DNA methylation or histone modifications can be restored using dCas9 protein fused or non-covalently bound to epigenetic effectors, derived from writers or erasers. Gene expression has been activated by DNA demethylation using Tet1, H3K27 acetylation by p300, or H3K4 trimethylation by PRDM9. The antagonistic effect can be achieved either by promoter methylation employing Dnmt3a, removal of a methyl group from H3K4me1/2 and H3K9me2 by LSD1 or deacetylation of H3K27ac by HDAC3.[56]
This entry is adapted from the peer-reviewed paper 10.3390/ijms21155314
References
- Ramaiya, K.J.; Harbour, J.W. Current management of uveal melanoma. Expert Rev. Ophthalmol. 2007, 2, 939–946.
- Jager, M.J.; Shields, C.L.; Cebulla, C.M.; Abdel-Rahman, M.H.; Grossniklaus, H.E.; Stern, M.-H.; Carvajal, R.D.; Belfort, R.N.; Jia, R.; Shields, J.A. Uveal melanoma. Nat. Rev. Dis. Primers 2020, 6, 1–25.
- Murray, T.G.; Boldt, H.C. Ocular Melanoma: Advances in Diagnostic and Therapeutic Strategies; Future Medicine: London, UK, 2014.
- Singh, A.D.; Turell, M.E.; Topham, A.K. Uveal melanoma: Trends in incidence, treatment, and survival. Ophthalmology 2011, 118, 1881–1885, doi:10.1016/j.ophtha.2011.01.040.
- Blum, E.S.; Yang, J.; Komatsubara, K.M.; Carvajal, R.D. Clinical management of uveal and conjunctival melanoma. Oncol. (Williston Park) 2016, 30, 29–43.
- Khoja, L.; Atenafu, E.; Suciu, S.; Leyvraz, S.; Sato, T.; Marshall, E.; Keilholz, U.; Zimmer, L.; Patel, S.; Piperno-Neumann, S. Meta-Analysis in metastatic uveal melanoma to determine progression free and overall survival benchmarks: an international rare cancers initiative (IRCI) ocular melanoma study. Ann. Oncol. 2019, 30, 1370–1380.
- Shields, C.L.; Ganguly, A.; Bianciotto, C.G.; Turaka, K.; Tavallali, A.; Shields, J.A. Prognosis of uveal melanoma in 500 cases using genetic testing of fine-needle aspiration biopsy specimens. Ophthalmology 2011, 118, 396–401.
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602.
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N. Mutations in GNA11 in uveal melanoma. New Engl. J. Med. 2010, 363, 2191–2199.
- Versluis, M.; de Lange, M.J.; van Pelt, S.I.; Ruivenkamp, C.A.; Kroes, W.G.; Cao, J.; Jager, M.J.; Luyten, G.P.; van der Velden, P.A. Digital PCR validates 8q dosage as prognostic tool in uveal melanoma. PLoS ONE 2015, 10.
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413.
- Kalirai, H.; Dodson, A.; Faqir, S.; Damato, B.; Coupland, S. Lack of BAP1 protein expression in uveal melanoma is associated with increased metastatic risk and has utility in routine prognostic testing. Br. J. Cancer 2014, 111, 1373–1380.
- Onken, M.D.; Worley, L.A.; Char, D.H.; Augsburger, J.J.; Correa, Z.M.; Nudleman, E.; Aaberg Jr., T.M.; Altaweel, M.M.; Bardenstein, D.S.; Finger, P.T. Collaborative Ocular Oncology Group report number 1: prospective validation of a multi-gene prognostic assay in uveal melanoma. Ophthalmology 2012, 119, 1596–1603.
- Field, M.G.; Harbour, J.W. Recent developments in prognostic and predictive testing in uveal melanoma. Curr. Opin. Ophthalmol. 2014, 25, 234.
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 2017, 32, 204–220. e215.
- Sharma, A.; Stei, M.; Fröhlich, H.; Holz, F.; Loeffler, K.; Herwig‐Carl, M. Genetic and epigenetic insights into uveal melanoma. Clin. Genet. 2018, 93, 952–961.
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428.
- Herlihy, N.; Dogrusöz, M.; Van Essen, T.H.; Harbour, J.W.; Van Der Velden, P.A.; Van Eggermond, M.C.; Haasnoot, G.W.; Van Den Elsen, P.J.; Jager, M.J. Skewed expression of the genes encoding epigenetic modifiers in high-risk uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1447–1458.
- Field, M.G.; Decatur, C.L.; Kurtenbach, S.; Gezgin, G.; Van Der Velden, P.A.; Jager, M.J.; Kozak, K.N.; Harbour, J.W. PRAME as an independent biomarker for metastasis in uveal melanoma. Clin. Cancer Res. 2016, 22, 1234–1242.
- Field, M.G.; Durante, M.A.; Decatur, C.L.; Tarlan, B.; Oelschlager, K.M.; Stone, J.F.; Kuznetsov, J.; Bowcock, A.M.; Kurtenbach, S.; Harbour, J.W. Epigenetic reprogramming and aberrant expression of PRAME are associated with increased metastatic risk in Class 1 and Class 2 uveal melanomas. Oncotarget 2016, 7, 59209.
- Venza, M.; Visalli, M.; Catalano, T.; Beninati, C.; Teti, D.; Venza, I. DSS1 promoter hypomethylation and overexpression predict poor prognosis in melanoma and squamous cell carcinoma patients. Hum. Pathol. 2017, 60, 137–146.
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068, doi:10.1038/nbt.1685.
- van der Velden, P.A.; Metzelaar-Blok, J.A.; Bergman, W.; Monique, H.; Hurks, H.; Frants, R.R.; Gruis, N.A.; Jager, M.J. Promoter hypermethylation: a common cause of reduced p16INK4a expression in uveal melanoma. Cancer Res. 2001, 61, 5303–5306.
- Van der Velden, P.A.; Zuidervaart, W.; Hurks, M.H.; Pavey, S.; Ksander, B.R.; Krijgsman, E.; Frants, R.R.; Tensen, C.P.; Willemze, R.; Jager, M.J. Expression profiling reveals that methylation of TIMP3 is involved in uveal melanoma development. Int. J. Cancer 2003, 106, 472–479.
- Zeschnigk, M.; Tschentscher, F.; Lich, C.; Brandt, B.; Horsthemke, B.; Lohmann, D.R. Methylation analysis of several tumour suppressor genes shows a low frequency of methylation of CDKN2A and RARB in uveal melanomas. Int. J. Genom. 2003, 4, 329–336.
- Maat, W.; van der Velden, P.A.; Out-Luiting, C.; Plug, M.; Dirks-Mulder, A.; Jager, M.J.; Gruis, N.A. Epigenetic inactivation of RASSF1a in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2007, 48, 486–490.
- Merhavi, E.; Cohen, Y.; Avraham, B.C.R.; Frenkel, S.; Chowers, I.; Pe’er, J.; Goldenberg-Cohen, N. Promoter methylation status of multiple genes in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4403–4406.
- Moulin, A.P.; Clément, G.; Bosman, F.T.; Zografos, L.; Benhattar, J. Methylation of CpG island promoters in uveal melanoma. Br. J. Ophthalmol. 2008, 92, 281–285.
- Calipel, A.; Abonnet, V.; Nicole, O.; Mascarelli, F.; Coupland, S.E.; Damato, B.; Mouriaux, F. Status of RASSF1A in uveal melanocytes and melanoma cells. Mol. Cancer Res. 2011, 9, 1187–1198.
- Pfeifer, G.P.; Yoon, J.-H.; Liu, L.; Tommasi, S.; Wilczynski, S.P.; Dammann, R. Methylation of the RASSF1A gene in human cancers. Biol. Chem. 2002, 383, 907–914.
- Maat, W.; Beiboer, S.H.; Jager, M.J.; Luyten, G.P.; Gruis, N.A.; van der Velden, P.A. Epigenetic regulation identifies RASEF as a tumor-suppressor gene in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1291–1298.
- Venza, M.; Visalli, M.; Beninati, C.; Biondo, C.; Teti, D.; Venza, I. Role of genetics and epigenetics in mucosal, uveal, and cutaneous melanomagenesis. Anti-Cancer Agents Med. Chem. (Former. Curr. Med. Chem. -Anti-Cancer Agents) 2016, 16, 528–538.
- Field, M.G.; Kuznetsov, J.N.; Bussies, P.L.; Cai, L.Z.; Alawa, K.A.; Decatur, C.L.; Kurtenbach, S.; Harbour, J.W. BAP1 loss is associated with DNA methylomic repatterning in highly aggressive Class 2 uveal melanomas. Clin. Cancer Res. 2019, 25, 5663–5673.
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719.
- Landreville, S.; Agapova, O.A.; Matatall, K.A.; Kneass, Z.T.; Onken, M.D.; Lee, R.S.; Bowcock, A.M.; Harbour, J.W. Histone deacetylase inhibitors induce growth arrest and differentiation in uveal melanoma. Clin. Cancer Res. 2012, 18, 408–416.
- Matatall, K.A.; Agapova, O.A.; Onken, M.D.; Worley, L.A.; Bowcock, A.M.; Harbour, J.W. BAP1 deficiency causes loss of melanocytic cell identity in uveal melanoma. BMC Cancer 2013, 13, 371.
- Kuznetsov, J.N.; Aguero, T.H.; Owens, D.A.; Kurtenbach, S.; Field, M.G.; Durante, M.A.; Rodriguez, D.A.; King, M.L.; Harbour, J.W. BAP1 regulates epigenetic switch from pluripotency to differentiation in developmental lineages giving rise to BAP1-mutant cancers. Sci. Adv. 2019, 5, eaax1738.
- Deng, S.; Calin, G.A.; Croce, C.M.; Coukos, G.; Zhang, L. Mechanisms of microRNA deregulation in human cancer. Cell Cycle 2008, 7, 2643–2646.
- Li, Y.; Jia, R.; Ge, S. Role of epigenetics in uveal melanoma. Int. J. Biol. Sci. 2017, 13, 426.
- Worley, L.A.; Long, M.D.; Onken, M.D.; Harbour, J.W. Micro-RNAs associated with metastasis in uveal melanoma identified by multiplexed microarray profiling. Melanoma Res. 2008, 18, 184–190.
- Radhakrishnan, A.; Badhrinarayanan, N.; Biswas, J.; Krishnakumar, S. Analysis of chromosomal aberration (1, 3, and 8) and association of microRNAs in uveal melanoma. Mol. Vis. 2009, 15, 2146.
- Venza, M.; Dell'Aversana, C.; Visalli, M.; Altucci, L.; Teti, D.; Venza, I. Identification of microRNA expression patterns in cutaneous and uveal melanoma cell lines. Tumori J. 2014, 100, 4–7.
- Sun, Q.; Cong, R.; Yan, H.; Gu, H.; Zeng, Y.; Liu, N.; Chen, J.; Wang, B. Genistein inhibits growth of human uveal melanoma cells and affects microRNA-27a and target gene expression. Oncol. Rep. 2009, 22, 563–567.
- Chen, X.; He, D.; Da Dong, X.; Dong, F.; Wang, J.; Wang, L.; Tang, J.; Hu, D.-N.; Yan, D.; Tu, L. MicroRNA-124a is epigenetically regulated and acts as a tumor suppressor by controlling multiple targets in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2248–2256.
- Chen, X.; Wang, J.; Shen, H.; Lu, J.; Li, C.; Hu, D.-N.; Da Dong, X.; Yan, D.; Tu, L. Epigenetics, microRNAs, and carcinogenesis: functional role of microRNA-137 in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1193–1199.
- Smolkova, B.; Horvathova, V.K.; Zmetakova, I.; Kalinkova, L.; Czanner, G.; Markova, A.; Furdova, A. Role of epigenetic deregulation in hematogenous dissemination of malignant uveal melanoma. Neoplasma 2018, 65, 840–854.
- Li, Z.; Yu, X.; Shen, J.; Jiang, Y. MicroRNA dysregulation in uveal melanoma: a new player enters the game. Oncotarget 2015, 6, 4562.
- Strub, T.; Ballotti, R.; Bertolotto, C. The “ART” of Epigenetics in Melanoma: From histone “Alterations, to Resistance and Therapies”. Theranostics 2020, 10, 1777.
- Nakade, S.; Yamamoto, T.; Sakuma, T. Cancer induction and suppression with transcriptional control and epigenome editing technologies. J. Hum. Genet. 2018, 63, 187–194.
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487.
- Akram, F.; Haq, I.U.; Ahmed, Z.; Khan, H.; Ali, M. CRISPR-Cas9, A Promising Therapeutic Tool for Cancer Therapy: A Review. Protein and Peptide Letters 2020. doi:10.2174/0929866527666200407112432.
- Moses, C.; Garcia-Bloj, B.; Harvey, A.R.; Blancafort, P. Hallmarks of cancer: The CRISPR generation. Eur. J. Cancer 2018, 93, 10–18.
- Wu, H.; Zhang, Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell 2014, 156, 45–68.
- 125. Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA methylation in the mammalian genome. Cell 2016, 167, 233–247. e217.
- Huang, Y.-H.; Su, J.; Lei, Y.; Brunetti, L.; Gundry, M.C.; Zhang, X.; Jeong, M.; Li, W.; Goodell, M.A. DNA epigenome editing using CRISPR-Cas SunTag-directed DNMT3A. Genome Biol. 2017, 18, 176.
- Syding, L.A.; Nickl, P.; Kasparek, P.; Sedlacek, R. CRISPR/Cas9 Epigenome Editing Potential for Rare Imprinting Diseases: A Review. Cells 2020, 9, 993.