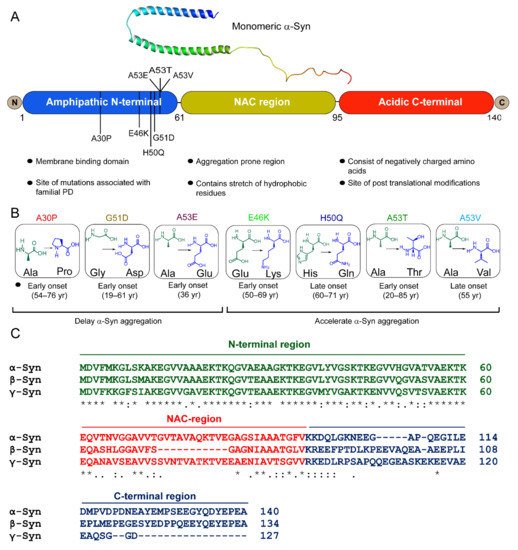
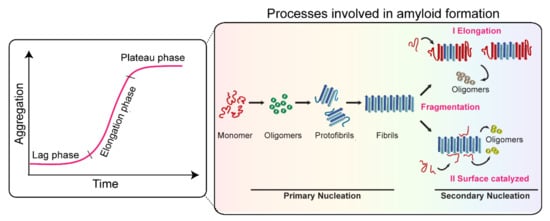
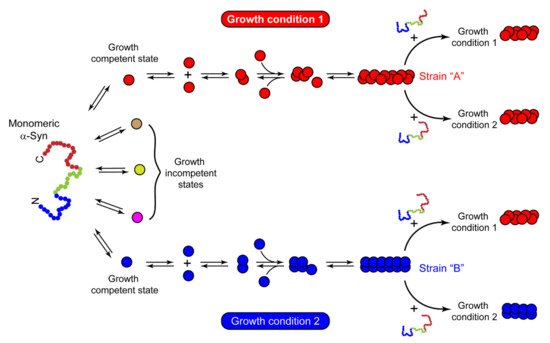
The structural and functional differences observed in recombinant strains can be validated by identifying and characterizing fibrils directly from synucleinopathy patient samples. The first evidence of a brain-derived strain came from a seminal study by Prusiner et al. [
270], which demonstrated that brain extracts from MSA are transmissible to transgenic mice and cells, resulting in abundant α-Syn pathology [
270]. In contrast, this was not observed using brain extracts from PD, suggesting that the PD-derived strain may differ from MSA [
270]. Then comes the question, what might lead α-Syn to adopt a different conformation in MSA or PD? In vitro, various solution conditions (like the presence or absence of salt) give rise to fibrils with different structural and functional properties [
29]. Similarly, α-Syn is also exposed to several microenvironments in vivo, affecting its aggregation [
271]. The dopaminergic neurons in PD and the oligodendrocytes affected in MSA belong to different cell lineages and have distinct cellular environments. Lee and co-workers demonstrated that distinct intracellular environments of two cell lines impart strain formation in MSA and PD [
34]. α-Syn fibrils derived from GCIs in oligodendrocytes (GCI-α-Syn) and LBs in neurons (LB-α-Syn) of diseased brains differ significantly and exhibit distinct seeding abilities [
34]. GCI-α-Syn strain is highly effective in seeding α-Syn aggregation compared to LB-α-Syn, thereby contributing to the aggressiveness of MSA [
34].
α-Syn aggregates have also been detected in biological fluids like cerebrospinal fluid (CSF) and plasma of PD patients [
272,
273]. α-Syn aggregation begins years before the onset of actual disease symptoms and, thus, the detection of these aggregates at early stages may enable the identification and characterization of a particular strain in these fluids. In this context, the amplification of α-Syn aggregates from brain extracts of PD and MSA patients using the protein misfolding cyclic amplification (PMCA) technique has been recently developed. This technique involves the amplification of misfolded proteins in vitro, in a manner similar to DNA amplification by PCR [
274]. It consists of alternate cycles of incubation and sonication, resulting in amyloid replication. First, the trace amount of amyloid is incubated with an excess of native protein to induce polymer growth. Then, the sample mixture is subjected to sonication, which will break down the fibrils, resulting in several nuclei. Each newly formed nucleus will then act as a seed in the next cycle and further induce the growth of fibrils. This way, after each cycle, the number of seeds will increase exponentially and will allow the detection of the minute amount of misfolded aggregates present at the beginning [
274]. Soto and co-workers used PMCA to amplify the α-Syn aggregates from the CSF of the patients diagnosed with PD and MSA [
52]. They found that PD- and MSA-derived fibrils exhibit different biophysical and biochemical properties and correspond to distinct conformational strains of α-Syn [
52]. Even α-Syn aggregates amplified from PD and MSA brain homogenates have been shown to exert variable toxicity and neurodegeneration in human dopaminergic neurons, reflecting different disease severity observed in PD and MSA patients due to different strains of α-Syn [
275]. These findings conclusively suggest that synucleinopathies can be distinguished based on the type of α-Syn strain present in the brain. However, the complexity in detecting aggregates arises when patient-to-patient heterogeneity is observed in the same disease. This heterogeneity in α-Syn aggregates amplified from PD patients’ brain extracts is greater than MSA brain extracts [
276]. Strohaker et al. reported that the fibrils derived from PD and MSA do not exhibit markedly distinct structural properties [
276], in contrast to findings reported by Soto and co-workers [
52]. The possible reason for the contrasting observations could be the differences in the PMCA protocols used by the two groups [
277]. Additionally, Strohaker et al. used a much smaller sample size than Soto’s group [
52,
276]. Other factors, like the genetic background of the patients, age of the selected patients, a load of α-Syn aggregates in different patients, presence of other components in the extracts, and region of the brain from where extraction was done, could also be responsible for these differences [
276]. Similar contradictions also exist in the field of AD pathology. Recent findings on brain-derived tau samples have suggested patient-to-patient heterogeneity in the tau fibril conformations exist within the same disease, AD [
278]. However, Goedert and colleagues observed the same type of tau conformation in all AD cases analyzed so far, suggesting that tau fibrils from a single disease (like AD or Pick’s disease) adopt a common structural fold [
218,
219]. Although the reasons and factors that drive this structural specificity in tauopathies are unclear, it could be due to multiple isoforms of tau, PTMs, interactions with other protein molecules, co-factors, etc. Recently, Scheres and Godert presented a hierarchal classification of tau fibrils from different tauopathies based on the folds of their filaments [
279]. Whether a similar classification exists for α-Syn fibrils isolated from synucleinopathy samples remains to be determined. Recently, a great effort has been made to solve the structure of α-Syn derived from the human brain by Schweighauser et al., using Cryo-EM [
280]. The group found that α-Syn filaments from the brain of DLB patients do not twist and are thinner than those derived from the brain of MSA patients [
280], consistent with the previous findings [
3]. The lack of twists in fibrils derived from DLB precluded the determination of 3D structure by cryo-EM and the differences in α-Syn fibrils derived from MSA and DLB patients were drawn based on two-dimensional class averaging [
280]. Although we need more high-resolution structures derived from synucleinopathy patients to reach a definite conclusion, the present reports certainly strengthen the claims on the existence of distinct fibril types of α-Syn. Moreover, the structures of α-Syn filaments from PD cases are not yet available, but solving them in the future can significantly help to understand the disease mechanism and generate therapeutic approaches against synucleinopathies.