Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Toxicology
Nanomaterials are now being used in a wide variety of biomedical applications. Medical and health-related issues, however, have raised major concerns, in view of the potential risks of these materials against tissue, cells, and/or organs and these are still poorly understood. These particles are able to interact with the body in countless ways, and they can cause unexpected and hazardous toxicities, especially at cellular levels. Therefore, undertaking in vitro and in vivo experiments is vital to establish their toxicity with natural tissues.
- nanomaterials
- nanoparticles toxicity
- nanomedicine
- toxicity assessment
- oxidative stress
- necrosis
- apoptosis
- biodistribution
- cell viability
- in vivo fate
1. Introduction
During the last 20 years, a significant growth in nanomaterial research has occurred, based on their numerous applications in medicine, photonics, and electronics [1,2,3,4,5]. Nanomaterials have been developed and used in a wide variety of commercial products, for example, electronic sensors, energy equipment, sun creams, and biomedical devices.
Toxicity research is important in order to fully understand the basic interaction of nanomaterials with natural tissues, because nanotechnology has a significant impact in the consumer and biomedical realms. One must understand the exact role of nanomaterials in vivo. When nanoparticles (NPs) are administrated into the human body, the effect of NP-loaded therapeutics, followed by their distribution into tissues (e.g., the kidneys, lung, and liver) must be precise. The clearance of nanomaterials can potentially occur through the pulmonary, hepatic, and renal systems. Thus, predicting long-term behavior becomes particularly difficult. NPs may aggregate in various organs and interact with off-target cells. They may degrade and be eliminated from the body due to their very small size [6]. There are, however, only a few studies examining whether nanomaterials are bio-compatible with natural organs, tissues, or cells. This review discusses the chemical, physical, and biological properties of various natural and synthetic nanomaterials, focusing on recent technologies related to the fabrication and processing of nanomaterials in different systems, emphasizing possible toxicity in in vitro and in vivo assays. We discuss the in vivo role of nanomaterials, including routes of administration, biodistribution, metabolism, routes of clearance, as well as blood biocompatability. The impacts of the physicochemical properties of nanomaterials and their toxicity are highlighted.
2. Mechanism of Toxicity
Nanoparticles can induce toxicity both in vitro and in vivo through various mechanisms. Examples of the most common mechanisms are oxidative stress, cell death mechanisms (apoptosis, autophagy, and necrosis), genotoxicity, and immunological responses. Each mechanism can lead to NP toxicity in a different pattern. The different mechanisms of NP toxicity are explained below.
2.1. Oxidative Stress
Oxidative stress is an imbalance between the production of reactive oxygen species (ROS) and antioxidant mechanisms [7] that can be explained as an increase in the generation of ROS or a decrease in antioxidants. A significant antioxidant found in animals, plants, and fungi is glutathione (GSH), which can halt losses caused by ROS. It is available in oxidized (GSSG) and reduced (GSH) forms. In usual healthy cases, most of the glutathione pool (about 90% of the total) is in a reduced state. An increased GSSG-to-GSH ratio indicates oxidative stress [8]. ROS can be triggered through various mechanisms, for example: (i) interaction with oxidative organelles such as mitochondria, (ii) involvement with redox active proteins, (iii) the chemical reaction of surface groups or coating from the NPs in the acidic environment, and (iv) the activation of different signaling routes via interaction with cell surface receptors [7,9]. In contrast, such reactions do not occur in the presence of all NPs, such as cerium oxide (CeO2). CeO2 NPs do not cause the formation of ROS, and they display a preserving effect against ROS damage both in vitro and in vivo [10,11]. Research shows that vitamin-C-conjugated NPs protect cells from oxidative stress at micromolar concentrations, and they induce cell death at millimolar concentrations [12]. The exact molecular target and chemical properties of oxidative stress, as well as the question of how it affects the modification of distinct biological procedures in cells subjected to engineered NPs (ENPs), have not yet been fully understood. Several studies postulate that reversible oxidative posttranslational modifications of protein cysteines via ROS and RNS show the basic mechanism of cell signaling that adjusts the protein functions of several cellular activities [13,14]. Protein S-glutathionylation (SSG) is a vital redox adjustment, altering apoptosis, mitochondrial metabolism, and transcription [15]. These SSG alterations are controlled by physiological properties. They can be repaired by glutaredoxin (Grx) enzymes [15,16]. Site-specific profiling of SSG modifications at a proteome-wide scale can be achieved through quantitative redox proteomics [17]. Using an SSG mechanism, Duan et al. assessed the modification of macrophage innate immune functions by ENPs via quantitative redox proteomics for the site-specific measurement of SSG alterations [18]. Three ENPs (silicon oxide (SiOx), Fe3O4, and cobalt oxide (II) (CoO)) were applied to stimulate disrupted macrophage function and cellular ROS, yielding low, moderate, and high propensity, respectively. SSG regulations indicate specific Cys residues and a wide range of redox-sensitive proteins corresponding to the total amount of cellular oxidative stress (CoO > Fe3O4 >> SiO2). ENPs that generate moderate and serious ROS show different pathways in response to SSG. Pathways modify protein stability and translation representative of the ER stress response. Proteins in phagocytosis, however, are highly susceptible to SSG in the presence of Fe3O4 [18]. ENPs trigger a subcytoxic degree of redox stress. ENP mitochondrial energetic pathways and classical stress responses are affected by SSG alternation moreso than redox stress caused by CoO.
2.2. Cell Death Mechanisms
This process is caused by cell replacement, or takes place when old cells are dying. Normally, it begins with organelle stress [19]. NPs can induce death-associated signaling via their interaction with various membranes or proteins in organelles. Specific proteins (receptors) involved in cell death are triggered by various NPs [20]. Cell death can be classified into three major mechanisms: apoptosis, autophagy, and necrosis.
2.2.1. Apoptosis
Apoptosis, alternatively known as type-1 cell death, is a process of programmed cell death that is critical for the development and preservation of tissue homeostasis [21]. Wang et al. demonstrated the inherent mechanism of caspase-dependent apoptosis with both intrinsic and extrinsic pathways on bone tumor impressions via selenium [22]. In vivo data analysis showed that selenium-doped hydroxyapatite NPs (Se-HNPs) cause tumor apoptosis to suppress tumor growth and reduce toxicity. Membrane integrity is inhibited during apoptosis (contrary to necrosis), since many molecules that are considered “eat me” signals in the plasma membrane of dying cells exist [23]. Pardo et al. clarified the molecular mechanism of apoptosis in mice for laser PTT [24]. Apoptosis induced via the intrinsic/mitochondrial pathway was regulated through the Bak and Bax pathways by activating the BH3-only protein Bid using laser-irradiated hot gold nanoprisms (NPRs). The NPR-mediated apoptosis obtained was related to caspase-3 and caspase-9 proteins. Another molecular mechanism was presented by Hou et al. for in vivo photodynamic therapy via the mitochondrial apoptosis pathway [25]. They applied an NIR light base on TiO2-modified upconversion NP (UCNP) core/shell nanocomposites (UCNPs@TiO2 NCs). NaYF4:Yb3+,Tm3+@NaGdF4:Yb3+ core/shell UCNPs effectively converted NIR light to UV emissions. The UCNPs@TiO2 NCs generate intracellular ROS in the presence of NIR irradiation, thus reducing the mitochondrial membrane potential to release cytochrome c in the cytosol, leading to the activation of caspase 3, and causing the apoptosis of the cancer cell. Instead of UV light, NIR-based UCNPs@TiO2 exhibits deeper tissue penetration and inhibits tumor growth more effectively [25].
2.2.2. Autophagy
Lin et al. have recently reported that tetrahedral DNA nanostructures (TDNs) containing a concentration of 250 nM improve autophagy and upregulate various autophagy-related proteins and genes [26]. TDNs are internalized through chondrocytes in the absence of other auxiliary agents. They focus on the cytoplasm of chondrocytes; however, only few single-strand DNAs (ssDNAs) are internalized by cells. TDNs increase autophagy via the PI3K/AKT/mTOR (mammalian target of rapamycin) pathway.
Several studies have revealed that NPs can disturb autophagy, which may lead to severe toxicological problems [27]. Additional research is required to establish the exact role of NPs in the induction of autophagy. This induction by a broad range of NPs (e.g., polymer [28], nanogel [29], and iron oxide NPs) has been studied [30]. Most of the examples cited are in vitro studies based on NP-induced autophagy that fail to imitate the complex in vivo physiological conditions. Zhou et al. evaluated QD-induced autophagy in living organismsm employing Caenorhabditis elegans (C. elegans) as the model organism [31]. They triggered autophagy in intestinal cells through the addition of QDs into the intestinal cells of C. elegans via oral feeding or microinjection. Figure 1 illustrates the real-time in situ tracking of autophagosome formation in live organisms.
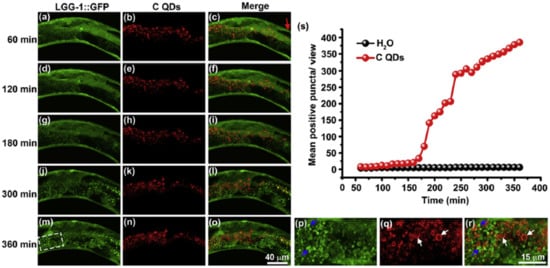
Figure 1. Real-time tracking of autophagosome formation in intestinal cells of C-QD-injected worms. (a–c), (d–f), (g–i), (j–l), and (m–o) are the fluorescent microscopy images of LGG-1::GFP and C QDs in intestinal cells of a C-QD-injected worm at 60 min, 120 min, 180 min, 300 min, and 360 min post-microinjection, respectively; (p–r) present enlarged views of the boxed areas in (m–o); blue arrows indicate the aggregated LGG-1::GFP; white arrows point to the internalized QDs; the red arrow in (c) indicates the injection site in the intestinal cells; (s) quantitative analysis of the aggregated LGG-1::GFP puncta subjected to C QDs or H2O treatment over 6 h. LGG-1 is the worm ortholog of the vacuolar protein Atg8/MAP-LC3). Reproduced with permission from [31], Elsevier, 2015.
This in vivo QD-induced autophagy is a defensive strategy of the organism aiming to degrade and recycle damaged cellular organelles. Fan et al. developed an autophagy regulation strategy based on folic-acid-modified N-doped carbon dots (FA-CDs) for visualized tumor therapy [32]. Stable FN-CDs exist in autophagic vacuoles in tumor cells. Their specific cellular uptake rate is a maximum of 93.40%. Cellular metabolism is influenced by FN-CDs that cause autophagy. Autophagic vacuoles are opened through autophagy inhibitors, and they release NP-activated signaling pathways, thereby killing tumor cells. This strategy shows therapeutic efficacy in 26 types of tumor cell lines. In vivo results indicate a greater efficiency (98%) than in traditional treatment (68%). Real-time in situ image tracking of the treatment has been reported through the use of FA-CDS.
2.2.3. Necrosis
Liu et al. reported that graphene oxide (GO) triggered necrosis in macrophages through the activation of Toll-like receptor 4 (TLR4), thus signaling autocrine TNF-α production [33]. The prevention of TLR4 signaling with a selective inhibitor almost completely inhibits cell death. GO-induced necrosis of macrophages is partially related to the RIPK1-RIPK3 complex necrosis associated with TNF-α induction. GO treatment leads to massive intracellular ROS generation. GO uptake into macrophages causes a reduction in the ability to perform phagocytosis and the disruption of the cytoskeleton. Zhang et al. reported that single-walled carbon-nanohorns (SNH) and conventional carbon nanotubes (CNT) induce cell death via necrosis and apoptosis [34]. They showed that CNT triggers 1.9–4.8-fold increases in necrosis compared to SNH, thereby indicating that SNH initiates significantly lower necrosis inducibility (Figure 2). Therefore, SNH and CNT trigger cell death through pyroptosis-mediated necrosis (pyroptosis is a commonly regulated necrosis pathway), and SNH always induces lower cell death than CNT via apoptosis or necrosis.
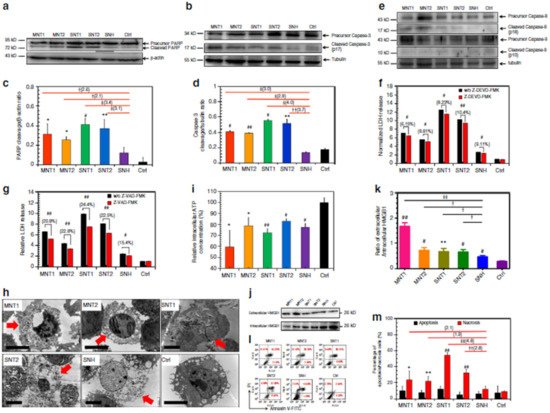
Figure 2. SNH induced less apoptosis and necrosis than CNT. (a,b) Western blot analyses of (a) PARP and (b) caspase-3 cleavages after different nanocarbon incubations. (c,d) Quantitative cleavage ratio measurements of (c) PARP and (d) caspase-3 in nanocarbon-incubated cells according to the integrated optic density (IOD) value, detected based on Western blot imaging (n = 3). (e) Western blot analyses of caspase-8 and caspase-9 cleavages after nanocarbon incubations. (f,g) Cytotoxicity detection of different nanocarbons with and without two caspase inhibitors, (f) Z-DEVD-FMK and (g) Z-VAD-FMK) (n = 4). (h) Transmission electron microscopy images of dead cells caused by different nanocarbons. Red arrows show the typical necrosis characteristics of cells. Scale bar: 5 μm. (i) Intracellular ATP detection after nanocarbon incubations (n = 4). (j) Immunoblot analysis of extracellular and intracellular HMGB1 after cellular incubations with different nanocarbons. (k) Quantitative ratio of extracellular HMGB1 to intracellular HMGB1 according to the IOD detection based on WB imaging (n = 3). (l) Flow cytometry analysis of cells based on the Annexin V/PI assay after nanocarbon incubations. (m) Quantitative comparison of apoptosis and necrosis caused by different nanocarbons detected using an apoptosis/necrosis assay kit (n = 4). In (c,d,f,g,i,k,m), data are presented as means ± s.d. Statistical significances were calculated by Student’s t-test. In (c,d,k), and (m), data were compared with control (Ctrl) and SNH groups separately. Versus Ctrl: * p < 0.05, ** p < 0.01, # p < 0.005, ## p < 0.001. Versus SNH: † p < 0.05, †† p < 0.01, ‡ p < 0.005, ‡‡ p < 0.001. The values in brackets denote the data ratios compared to SNH group. In f, g, data were compared with no-inhibitor-added groups for each type of nanocarbons: # p < 0.005; ## p < 0.001. Reproduced with permission from [34], Nature, 2018. Abbreviations: PARP, poly-ADP-ribose polymerase; Z-DEVD-FMK, specific caspase-3 inhibitor; Z-VAD-FMK, pan-caspase inhibitor; MNT, multi-walled carbon nanotubes, SNT, single-walled carbon nanotubes.
Among programmed cell death processes, necrosis is considered the most common cellular response during tumor ablation performed through NP-mediated PTT [35]. In general, uncontrolled necrosis causes the release of intracellular constituents into the extracellular milieu. Therefore, induced responses may generate a harmful knock-on impact by damaging adjacent tissue, triggering tumor growth, etc. [36]. Loss of membrane integrity occurs particularly in necrotic cells. However, when considering in vitro environments, apoptotic cells also suffer from membrane integrity loss (secondary necrosis) in the absence of phagocytic cells. This behavior occurs later than during primary necrosis.
2.3. Genotoxicity
The destruction of genetic material in a cell due to toxicity is usually referred to as genotoxicity [37]. This includes DNA or chromosomal damage, including gene mutations, chromosome rearrangements, and breaks [38] using sophisticated signal transduction pathways [39]. Nanomaterials may influence genotoxicity because of their unique properties, for example, their very small size and broad surface area. Several factors are involved in the interaction of genetic materials with CNTs, e.g., similarity in size to that of microtubules, the capability of CNTs to penetrate the nuclear membrane, and the high affinity of SWCNTs to the G-C-rich areas in DNA sequences [40]. Reportedly, MWCNTs and SWCNTs triggered DNA damage in vitro in mesothelial cells [41]. NPs generate genotoxicity directly (primary genotoxicity) or indirectly (secondary genotoxicity). Inflammatory responses and oxidative stress cause secondary genotoxicity; primary genotoxicity may occur through an interaction of NPs with DNA or the mitotic apparatus [42]. Indirect genotoxicity of NPs can occur via the production of ROS through inflammatory cells. The resultant ROS can interact with DNA, ending in DNA oxidation, breakage, and/or lipid-peroxidation-mediated DNA adducts [43].
Genotoxicity can be detected in vitro by the Ames test, comet assay, and the chromosome aberration test, and can also be detected in vivo (rodent carcinogenicity, chromosome aberration, and mutation of endogenous genes). These assays are generally based on the detection of induced damages. Numerous NPs are reportedly genotoxic, such as polymeric and engineered NPs [44,45]. Many human cells have been used to evaluate the genotoxicity of CNTs. These include fibroblasts and RAW264.7 macrophages, normal lung epithelial BEAS-2B and SAEC cells, normal and malignant mesothelial cells, as well as malignant alveolar epithelial A549 cells. They show that high doses (>50 to 100 μg mL−1) of MWCNT and SWCNT induce genotoxicity. CNTs disrupt their dynamics and functions by imitating microtubules [46]. The Ames test indicates a sensitivity lower than that of micronucleus assays in terms of the genotoxicity assessment of AgNPs in human lymphoblastoid (TK6) cells [47]. One can attribute lesser sensitivity of the Ames test, which employs bacterial cells to evaluate mutagenicity, to non-endocytic bacterial effects. For this reason, the Ames test is not suitable for specific NPs [48]. The micronucleus assay can also yield false results because of reagent interference (i.e., cytochalasin B) with the cellular uptake of NPs [49]. DNA damage caused by NPs has also been measured by means of 8-oxoguanine (8-oxo-dG) ELISA [50]. This assay attains high throughput with a 96-well plate design; however, it measures only a biomarker of DNA oxidation, 8-oxo-dG, and does not reveal other DNA damage such as alkali-sensitive sites, double- and single-strand breaks, and abasic sites. The comet assay is the most common test for assessing the genotoxicity of nanomaterials. It has low reproducibility and throughput because of sample-to-sample variation (one sample displayed different results when tested on two separate glass slides) [51]. Watson et al. introduced a high-throughput screening (HTS) assay based on the CometChip approach that provides assessments of abasic sites, alkali-sensitive sites, and single-stranded DNA breaks in TK6 and adherent Chinese hamster ovary (H9T3) cells [52]. The researchers noted dose-dependent increases in cytotoxicity and DNA damage through the exposure to engineered nanoparticles (ENPs) (Ag, CeO2, Fe2O3, SiO2, and ZnO) at a dose range of 5, 10, and 20 μg/mL. The genotoxicity of ENPs in TK6 cells at 4 h and in H9T3 cells at 24 h was ZnO > Ag > Fe2O3 > CeO2 > SiO2, and Ag > Fe2O3 > ZnO > CeO2 > SiO2, respectively (Figure 3).
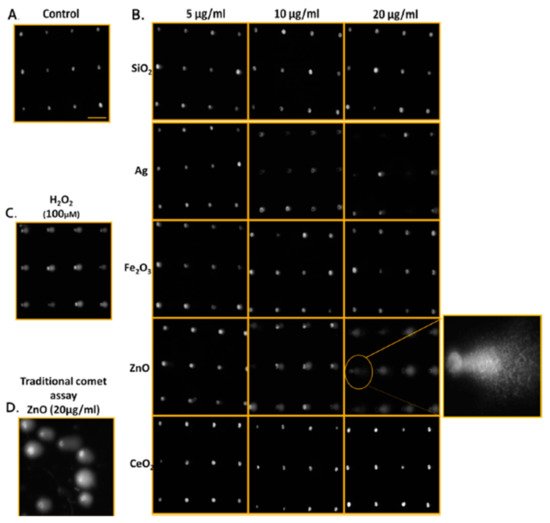
Figure 3. Qualitative images of nanoparticle-mediated ENP DNA damage in TK6 cells using both CometChip and standard comet assays. (A) Media-treated control cells. (B) TK6 cells were exposed to industrially relevant ENPs at concentrations of 5, 10, and 20 μg/ml for 4 h and evaluated using CometChip technology. The expanded view illustrates the morphology of the comet structure induced from 4 h exposure of zinc oxide ENP in TK6, revealing significant DNA damage. (C) Positive control cells treated with H2O2 (100 μM) for 20 min. (D) Traditional comet assay of TK6 cells treated with ZnO (20 μg/ml) for 4 h for comparison to CometChip qualitative assessments. Horizontal scale bar represents 100 μm. Reproduced with permission from [52], American Chemical Society, 2014.
2.4. Immune Response
A wide range of NPs can stimulate, suppress, and/or elicit no immune response. Interaction with immune responses is associated with physiochemical properties that include shape, size, and charge [53]. NPs are able to adsorb, encapsulate, and/or chemically immobilize antigens within their surfaces, thus resulting in controlled release and delivery [54,55]. Niikura et al. demonstrated that various shapes of AuNPs modified with Nile virus envelope (E) protein cause cytokine secretion behavior in dendritic cells. Cubic and spherical AuNPs lead to the secretion of pro-inflammatory cytokines at high levels, but rod AuNPs cause the secretion of inflammasome-related cytokines interleukin (IL) 1β and IL-18 [56]. Long-aspect-ratio ENPs, (e.g., CNTs and nanowires) induce the activation of the inflammasome (secondary to oxidative stress) and induce a shape-dependent effect at the lysosomal level [57]. NPs may also activate cell and humoral immune responses through interaction with cell receptors and plasma proteins. They can also be internalized via mammalian cells. NPs can deliver sufficient antigens to targeted sites and enhance immune responses via their favorable bioavailability and cellular uptake, thereby reducing adverse effects [58].
Nanomaterials can be designed as vaccines to improve immune responses by reducing the unwanted activation or suppression of immune systems. This is suitable for infectious diseases and the immunotherapy of tumors [59]. Nanovaccines can regulate their hydrophilicity, shape, antigen loading, surface potential, and scale through various fabrication procedures and materials [59]. They promote long-term immune responses via adhesion, mixing, conjugation, and encapsulation of the antigen with NPs for vaccine delivery [60]. Pulendran et al. developed synthetic polymer NPs loaded with hemagglutinin (HA) from Toll-like receptor ligands and avian influenza H5N1. This is effective against swine influenza virus strains and lethal avian influenza in mice. It also causes robust immunity against H1N1 influenza [61]. Despite these NP-based vaccines, there are examples indicating that non-vaccine nanomaterials can also induce immune response [62]. Zhai et al. designed crosslinked (X-NPs) and non-crosslinked self-assembled NPs (NX-NPs) for antimetastasis and antiproliferation of melanoma [63]. Chondroitin sulfate (CS)-adipic dihydrazide (ADH)-chlorin e6 (Ce6)-lipoic acid (LA) loaded docetaxel (DTX) works as a redox/enzyme/ultrasound. Response conjugates form self-assembled NPs in the presence of aqueous solutions that are suitable for combining sonodynamic therapy (SDT) and chemotherapy. X-NPs induce the generation of ROS, leading to mitochondrial damage, followed by cell apoptosis via the mitochondria-caspase pathway. CD44 nanoscale distribution and targeted properties initiate the successful tumor homing ability of NPs, thereby causing potential antimetastasis and antitumor efficiencies, with declined expression of tumorous uPA and COX-2 proteins. The SDT of X-NPs triggers an immunological response via cell death. It causes the release of tumor-associated antigens.
3. Toxicity Assessment of Nanomaterials
3.1. In Vitro Characterization
The in vitro evaluation of biocompatibility with blood components is a necessary part of early preclinical development. Many research projects have reported NPs’ hemolytic properties. It is probable that the biological activity of NPs depends on physicochemical parameters that are not routinely considered in toxicity screening studies. Physicochemical properties may be important in comprehending the toxic effects of test materials. These properties include particle size and size distribution, shape, crystal structure, chemical composition, agglomeration state, surface area, surface chemistry, and charge, as well as porosity. In vitro techniques allow specific biological and mechanistic pathways to be isolated and tested under controlled conditions that are not feasible in in vivo tests. Various tests have been suggested for portal-of-entry toxicity for lungs, skin, and mucosal membranes and to target organ toxicity for the endothelium, liver, nervous system, heart, blood, spleen, and kidney. Non-cellular assessment of NPs’ durability, protein interactions, complement activation, and pro-oxidant activity have also been considered. Section 3.1 highlights the most basic in vitro characterizations.
3.1.1. Electron Microscopy
Horvath et al. examined the in vitro cytotoxicity of boron nitride nanotubes (BNNTs) using SEM [64]. Nanotubes attached to the surface of exposed cells can cause toxicity by activating signaling pathways. SEM, therefore, has been applied to study the distribution of BNNTs on the surface of macrophages (RAW 264.7) and epithelial cells (A549). BNNT-treated A549 cells reveal no significant morphological changes, whereas many nanotubes bind to the cell surface in BNNT-treated macrophages. SEM indicates the ultrastructural modification of the A549 epithelial cell surface in the response of ENMs. The biocompatibility and dynamical processes of the interaction of nanomaterials with macrophage cells dosed with metal oxide NPs (CeO2, TiO2 and ZnO) were examined in another study through field emission scanning electron microscopy (FE-SEM) [65]. FE-SEM confirmed the interaction and the exact details of the internalization, adsorption, and ultrastructural location of NPs via the main constituents of the innate immune system. FE-SEM offers new insights into the biocompatibility and biodistribution of NPs with immune cells. SEM was also used to determine tumor margins based on the actual visualization of anti-AuNR-epidermal growth factor receptors (EGFRs) [66]. The distribution of EGFRs in and around tumor cells was evaluated using air SEM (airSEM), a highly sensitive form of microscopy for biological samples. AirSEM provides localized and actual visualization of the AuNR on tissues. The distance between healthy and tumor regions is within a radius of ~1 mm (Figure 4a).
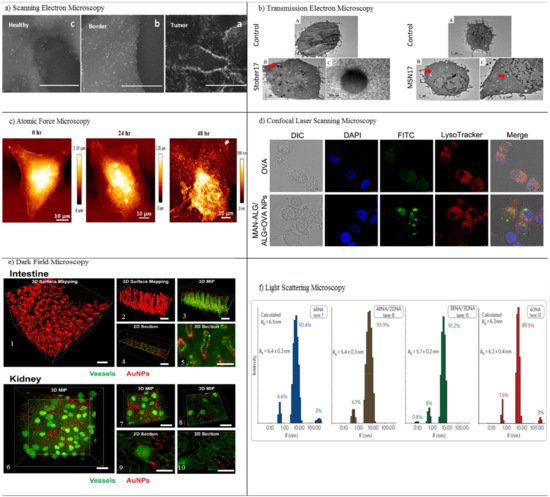
Figure 4. In vitro characterization methods. (a) Scanning electron microscopy (SEM) images of the different sites (a–c) in the oral SCC slide, in a nanometric resolution. The GNRs appear as bright rods. The nanoparticles’ concentration gradually decreases from the tumor to the healthy sites. Reproduced with permission from [66], American Chemical Society, 2016. (b) Transmission electron microscopy (TEM) of RAW264.7 cells treated with media (control) or treated with different concentrations of porous and nonporous SNPs for 4 h. The SNPs were taken up by cells and localized inside vesicles. The dose-dependent increase of the cellular association of SNPs in both types of nanoparticles is visualized. Red arrows indicate particles inside cells. Particles were not observed inside the nucleus. Reproduced with permission from [69], Elsevier, 2017. (c) Atomic force microscopy (AFM) imaging of CUR-AuNCs treated with HeLa cells in different intervals of time, 0, 24, and 48 h. Reproduced with permission from [74], American Chemical Society, 2018. (d) Confocal laser scanning microscopy (CLSM) imaging of DCs after incubation with free OVA and MAN-ALG/ALG=OVA NPs. Reproduced with permission from [77], Elsevier, 2017. (e) 3D dark field microscopy of AuNPs in mouse intestine and kidney tissues, with (1) surface mapping of the 3D image of intestinal tissue containing 50-nm AuNPs, showing the morphology of villi. (2) Smaller segment of the image from (1). (3) 3D maximum intensity projection (MIP) of the same region of intestinal tissue showing the arrangement of blood vessels and the distribution of AuNPs. (4) Position of the 2D section in (5) showing the distribution of AuNPs within a single villus. (6, 7) 3D maximum intensity projection of blood vessels and AuNPs within kidney tissue with brightly stained glomeruli visible. (8) Location of 2D sections of (9) and (10) showing the local distribution of AuNPs within and around a glomerulus. Reproduced with permission from [81], Royal Society of Chemistry, 2017. (f) Light scattering microscopy of size histograms for 6-stranded cubes. Reproduced with permission from [85], Nature, 2010.
Transmission electron microscopy (TEM) utilizes electrons transmitted through a sample to form images. TEM reveals considerable information for in vitro NP localization and uptake through the visualization of NPs’ locations in a cell/tissue, together with other techniques [67]. Both SEM and TEM are highly suitable for the characterization of nanomaterials before and after administration within samples. Several drawbacks limit their application: long sample preparation time with a low throughput, limited use of biological samples with thickness of 50–100 nm, and structural damage to the sample during preparation. Even so, TEM has been applied as a method in the cellular localization of various NPs [67,68]. It is also capable of detecting morphological changes on the cells caused by NPs. Ghandehari and co-workers utilized TEM for the cellular internalization of silica nanoparticles (SNPs) composed of nonporous spherical (Stöber) SNPs with diameters of 46 ± 4.9 nm (Stöber50), 432 ± 18.7 nm (Stöber500), and mesoporous spherical (MSN) particles with a diameter of 466 ± 86 nm (MSN500) [69]. The SNPs were localized inside vesicles situated in the cytosol of macrophages. TEM imaging shows that the number of Stöber500 particles attached to the outer part of cell was higher than that of MSN500 particles. Compared to Stöber500 SNPs, the higher internalization of MSN500 is sub-cytotoxic and causes enlargement of macrophage cells. Both nonporous and mesoporous spherical SNPs were applied to study the gene expression of cells. Stöber SNPs have a minor effect on gene expression. MSN, however, can change gene transcription without exhibiting any acute toxicity (Figure 4b). Scanning transmission electron microscopy (STEM) is a modified type of TEM that forms images by utilizing the combination of suitable detectors. Valiyaveettil et al. used STEM to investigate the biodistribution of silver NPs. 100 mg mL−1 of silver NPs was treated with cancer cells [70]. The presence of silver NPs in the mitochondria and nucleus was imaged by STEM; the direct effect of silver NPs in DNA damage and mitochondrial toxicity was confirmed. Peckys et al. applied the STEM method to provide information about the intracellular uptake of 30-nm AuNPs in live fibroblast cells (COS-7) in a microfluidic chamber [71]. Information about the distribution of AuNPs in pristine cells was obtained, since the cells are alive at the onset of STEM. That information includes the number of vesicles in the cluster, the vesicle diameter, and the number of detected AuNPs per 0.15 × 0.15 μm2 of the image.
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics13101615
This entry is offline, you can click here to edit this entry!